

Mycobacterium abscessus pathogenesis identified by phenogenomic analyses
- PMID: 36008617
- PMCID: PMC9418003
- DOI: 10.1038/s41564-022-01204-x
Mycobacterium abscessus pathogenesis identified by phenogenomic analyses
Abstract
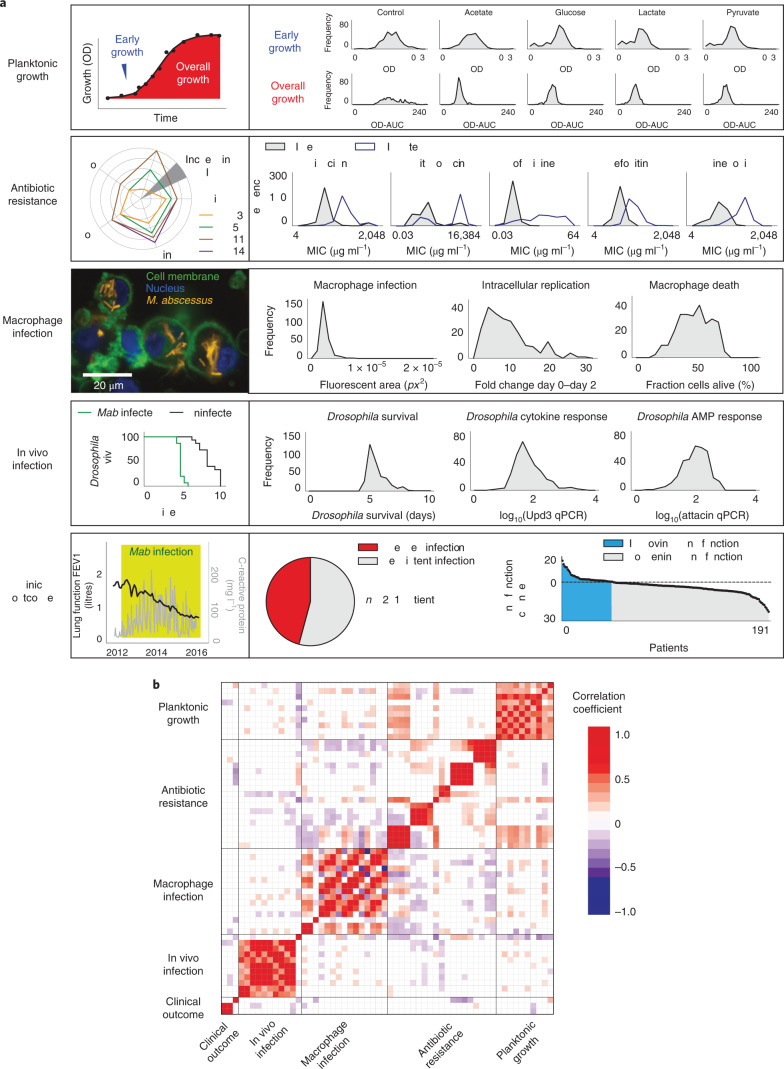
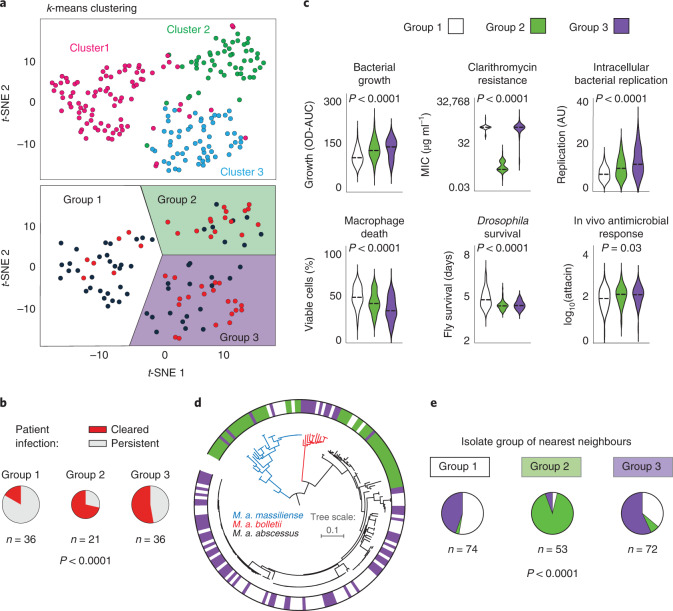
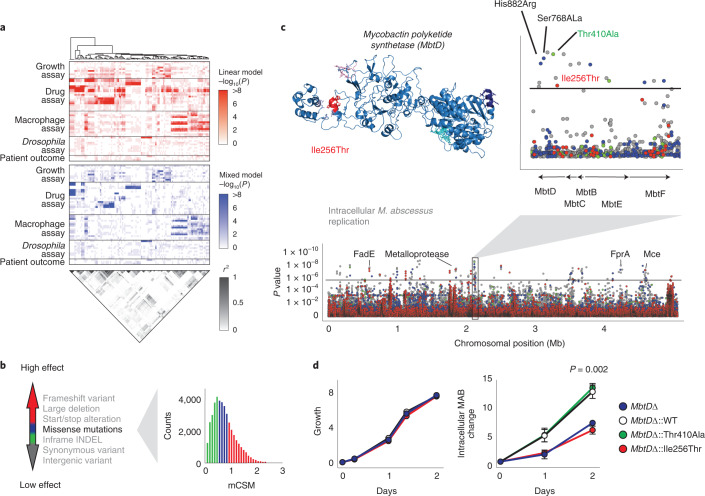
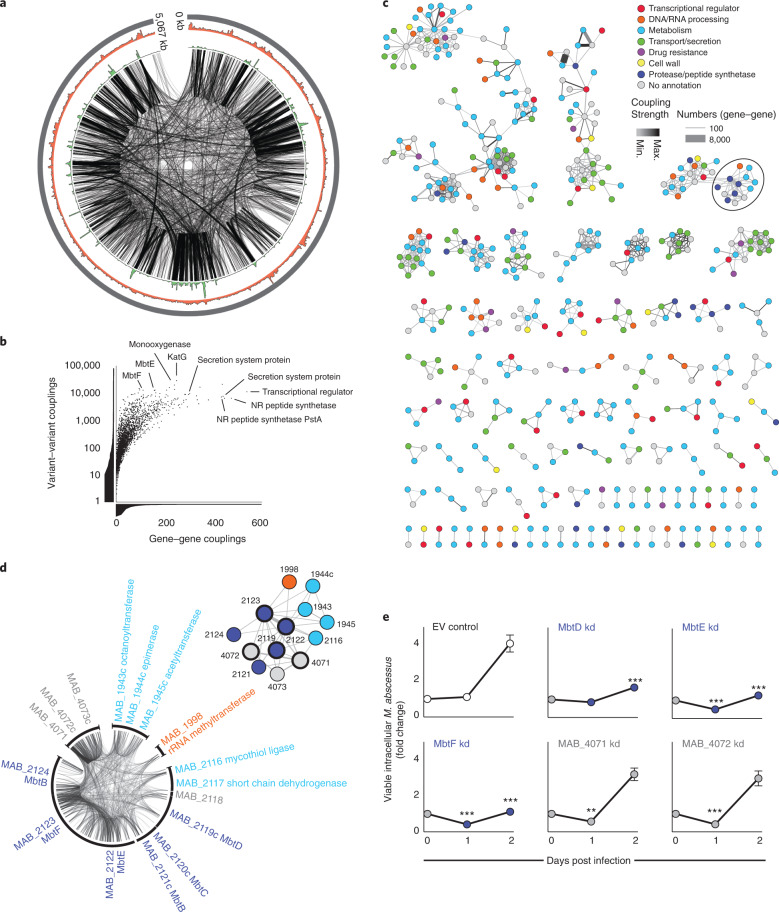
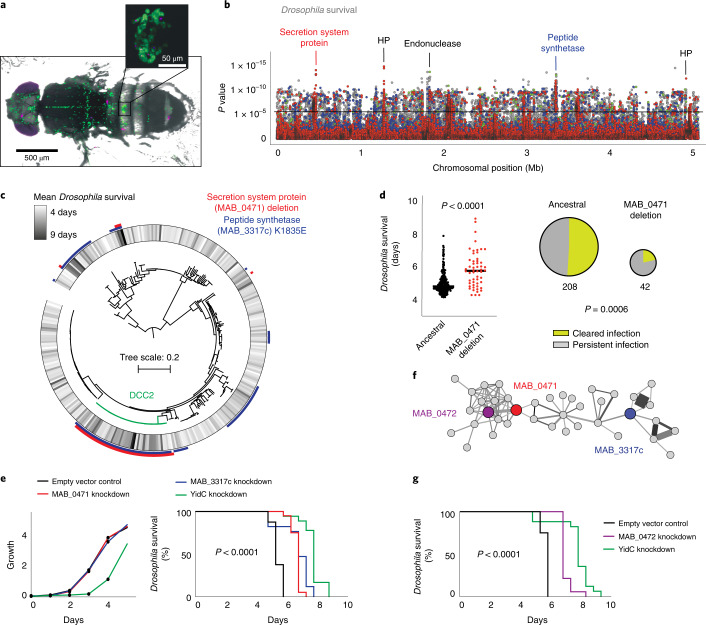
The medical and scientific response to emerging and established pathogens is often severely hampered by ignorance of the genetic determinants of virulence, drug resistance and clinical outcomes that could be used to identify therapeutic drug targets and forecast patient trajectories. Taking the newly emergent multidrug-resistant bacteria Mycobacterium abscessus as an example, we show that combining high-dimensional phenotyping with whole-genome sequencing in a phenogenomic analysis can rapidly reveal actionable systems-level insights into bacterial pathobiology. Through phenotyping of 331 clinical isolates, we discovered three distinct clusters of isolates, each with different virulence traits and associated with a different clinical outcome. We combined genome-wide association studies with proteome-wide computational structural modelling to define likely causal variants, and employed direct coupling analysis to identify co-evolving, and therefore potentially epistatic, gene networks. We then used in vivo CRISPR-based silencing to validate our findings and discover clinically relevant M. abscessus virulence factors including a secretion system, thus illustrating how phenogenomics can reveal critical pathways within emerging pathogenic bacteria.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Phenogenomics of Mycobacterium abscessus.Nat Microbiol. 2022 Sep;7(9):1325-1326. doi: 10.1038/s41564-022-01217-6. Nat Microbiol. 2022. PMID: 36008618 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
