

Lipid Metabolism in Glioblastoma: From De Novo Synthesis to Storage
- PMID: 36009491
- PMCID: PMC9405736
- DOI: 10.3390/biomedicines10081943
Lipid Metabolism in Glioblastoma: From De Novo Synthesis to Storage
Abstract
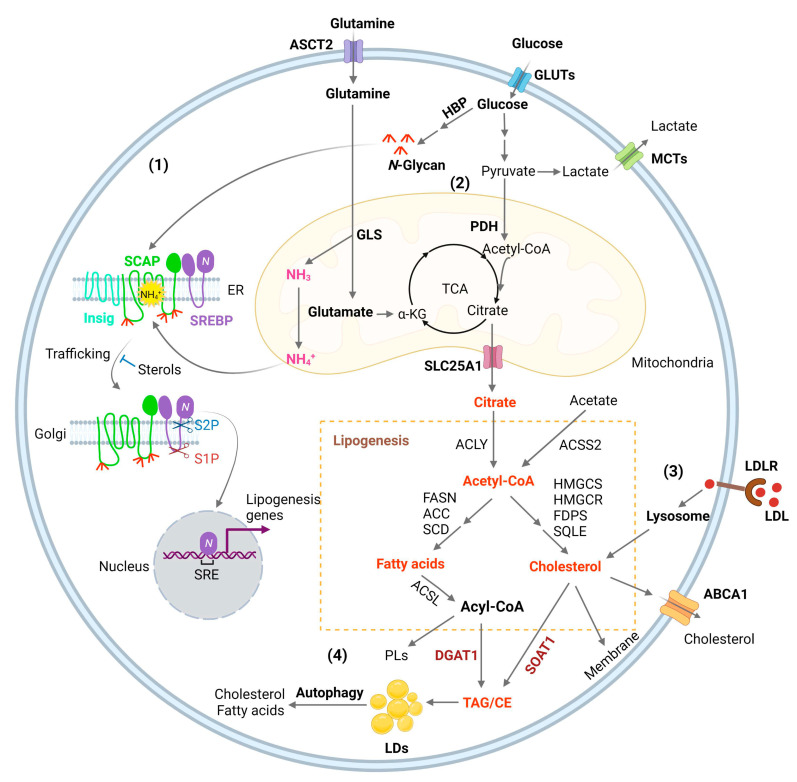
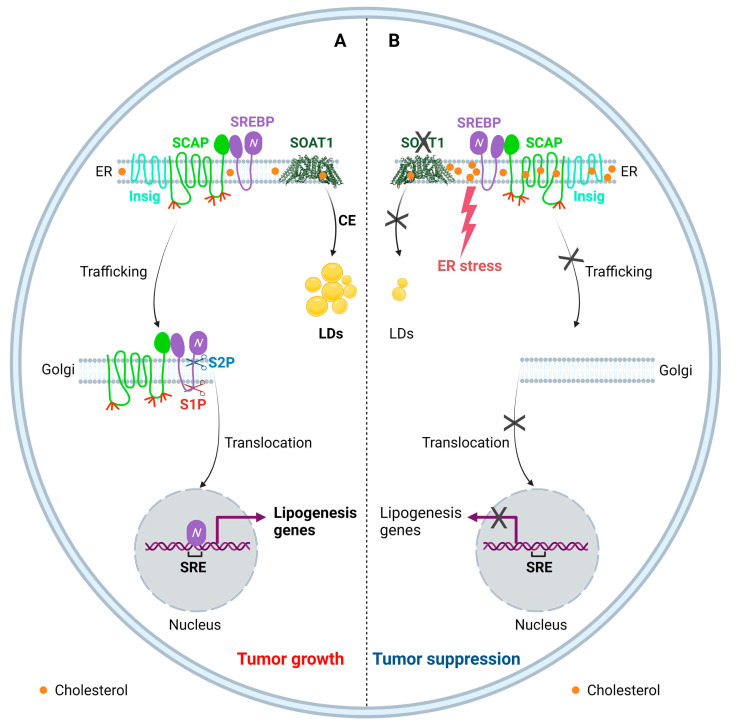
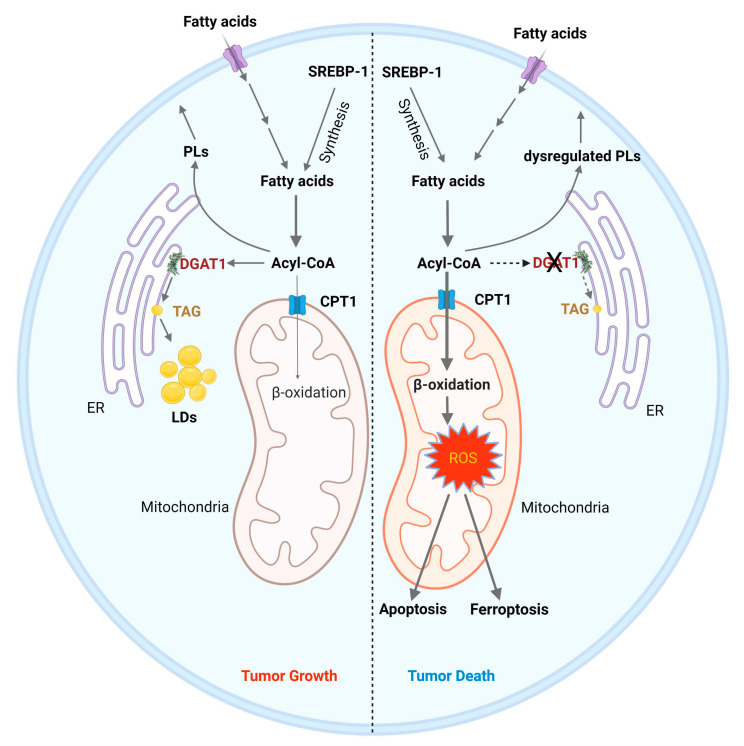
Glioblastoma (GBM) is the most lethal primary brain tumor. With limited therapeutic options, novel therapies are desperately needed. Recent studies have shown that GBM acquires large amounts of lipids for rapid growth through activation of sterol regulatory element-binding protein 1 (SREBP-1), a master transcription factor that regulates fatty acid and cholesterol synthesis, and cholesterol uptake. Interestingly, GBM cells divert substantial quantities of lipids into lipid droplets (LDs), a specific storage organelle for neutral lipids, to prevent lipotoxicity by increasing the expression of diacylglycerol acyltransferase 1 (DGAT1) and sterol-O-acyltransferase 1 (SOAT1), which convert excess fatty acids and cholesterol to triacylglycerol and cholesteryl esters, respectively. In this review, we will summarize recent progress on our understanding of lipid metabolism regulation in GBM to promote tumor growth and discuss novel strategies to specifically induce lipotoxicity to tumor cells through disrupting lipid storage, a promising new avenue for treating GBM.
Keywords: DGAT1; SOAT1; SREBP-1; cholesterol; fatty acids; glioblastoma; lipid droplets; lipotoxicity.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
