

The Cell Autonomous and Non-Cell Autonomous Aspects of Neuronal Vulnerability and Resilience in Amyotrophic Lateral Sclerosis
- PMID: 36009818
- PMCID: PMC9405388
- DOI: 10.3390/biology11081191
The Cell Autonomous and Non-Cell Autonomous Aspects of Neuronal Vulnerability and Resilience in Amyotrophic Lateral Sclerosis
Abstract
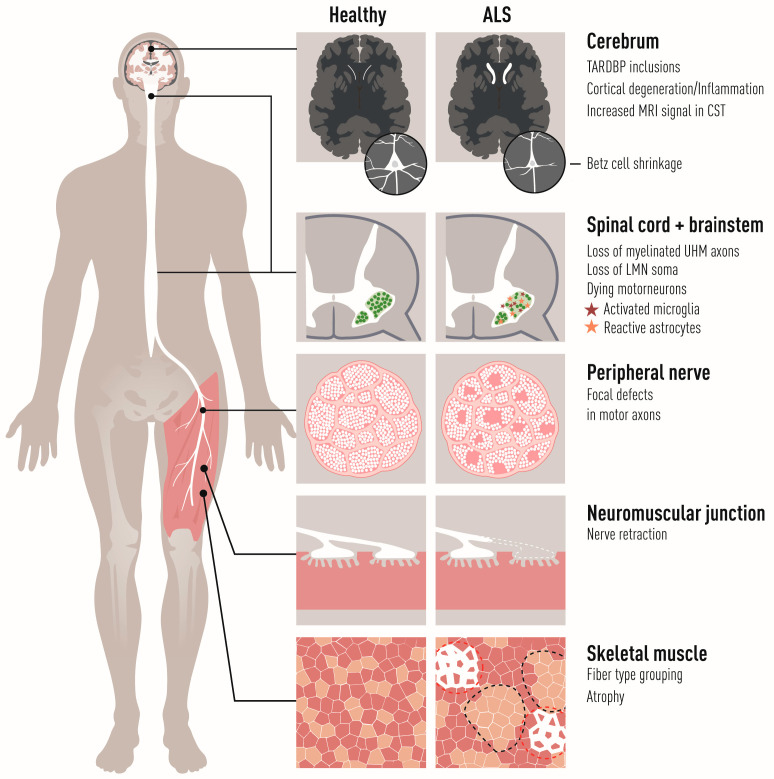
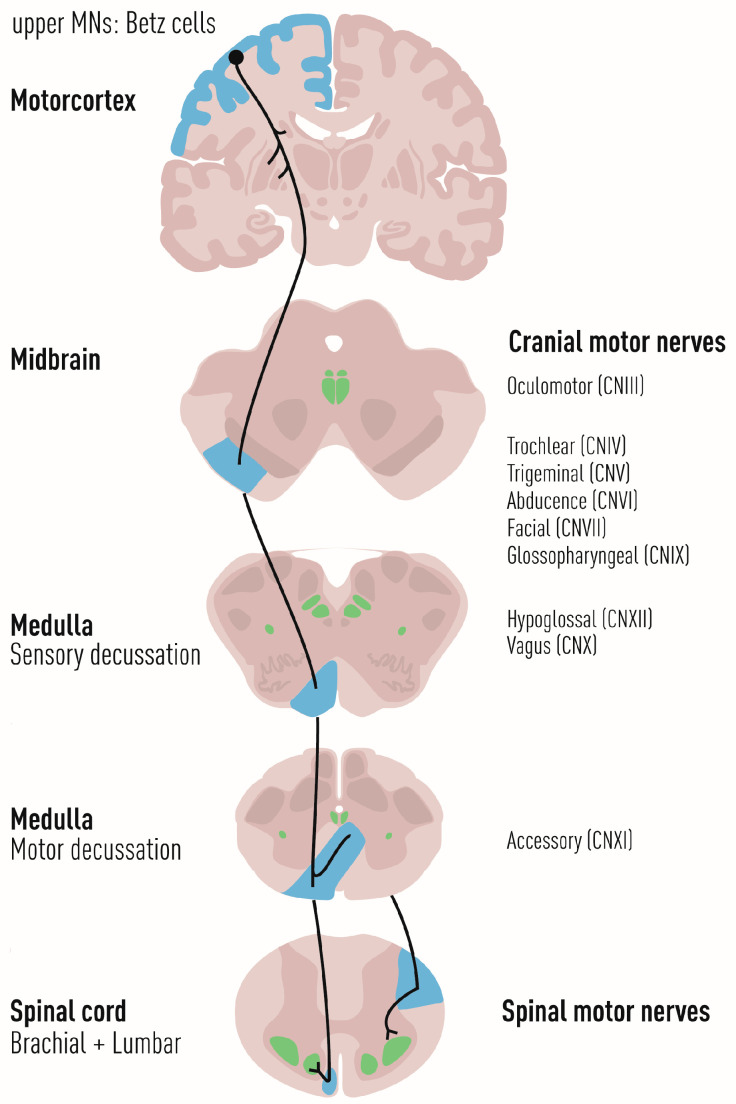
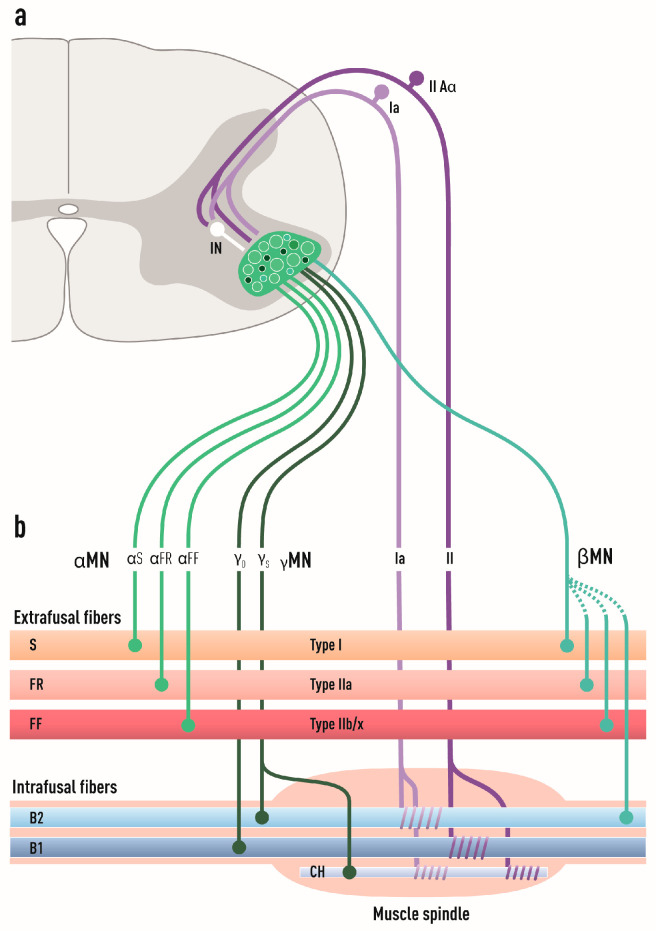
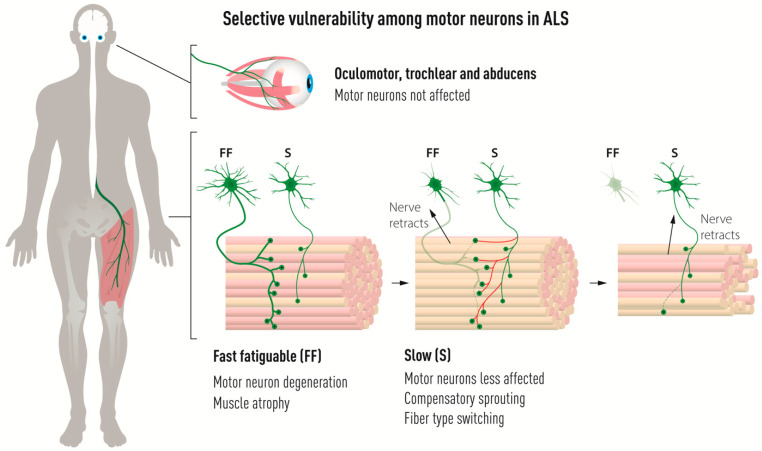
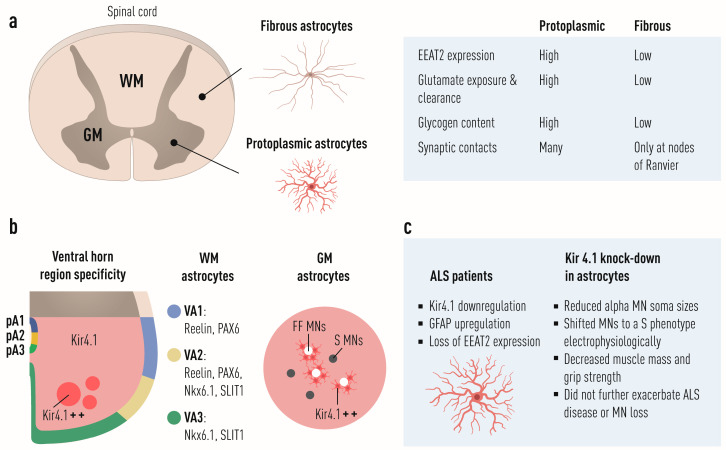
Amyotrophic lateral sclerosis (ALS) is defined by the loss of upper motor neurons (MNs) that project from the cerebral cortex to the brain stem and spinal cord and of lower MNs in the brain stem and spinal cord which innervate skeletal muscles, leading to spasticity, muscle atrophy, and paralysis. ALS involves several disease stages, and multiple cell types show dysfunction and play important roles during distinct phases of disease initiation and progression, subsequently leading to selective MN loss. Why MNs are particularly vulnerable in this lethal disease is still not entirely clear. Neither is it fully understood why certain MNs are more resilient to degeneration in ALS than others. Brain stem MNs of cranial nerves III, IV, and VI, which innervate our eye muscles, are highly resistant and persist until the end-stage of the disease, enabling paralyzed patients to communicate through ocular tracking devices. MNs of the Onuf's nucleus in the sacral spinal cord, that innervate sphincter muscles and control urogenital functions, are also spared throughout the disease. There is also a differential vulnerability among MNs that are intermingled throughout the spinal cord, that directly relate to their physiological properties. Here, fast-twitch fatigable (FF) MNs, which innervate type IIb muscle fibers, are affected early, before onset of clinical symptoms, while slow-twitch (S) MNs, that innervate type I muscle fibers, remain longer throughout the disease progression. The resilience of particular MN subpopulations has been attributed to intrinsic determinants and multiple studies have demonstrated their unique gene regulation and protein content in health and in response to disease. Identified factors within resilient MNs have been utilized to protect more vulnerable cells. Selective vulnerability may also, in part, be driven by non-cell autonomous processes and the unique surroundings and constantly changing environment close to particular MN groups. In this article, we review in detail the cell intrinsic properties of resilient and vulnerable MN groups, as well as multiple additional cell types involved in disease initiation and progression and explain how these may contribute to the selective MN resilience and vulnerability in ALS.
Keywords: amyotrophic lateral sclerosis; neurodegeneration; neurodegenerative cell metabolism; neuron.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in reviewing the literature or in the writing of the manuscript.
Figures
References
-
- Riva N., Clarelli F., Domi T., Cerri F., Gallia F., Trimarco A., Brambilla P., Lunetta C., Lazzerini A., Lauria G., et al. Unraveling gene expression profiles in peripheral motor nerve from amyotrophic lateral sclerosis patients: Insights into pathogenesis. Sci. Rep. 2016;6:39297. doi: 10.1038/srep39297. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
