

The Interaction of Human Papillomavirus Infection and Prostaglandin E2 Signaling in Carcinogenesis: A Focus on Cervical Cancer Therapeutics
- PMID: 36010605
- PMCID: PMC9406919
- DOI: 10.3390/cells11162528
The Interaction of Human Papillomavirus Infection and Prostaglandin E2 Signaling in Carcinogenesis: A Focus on Cervical Cancer Therapeutics
Abstract
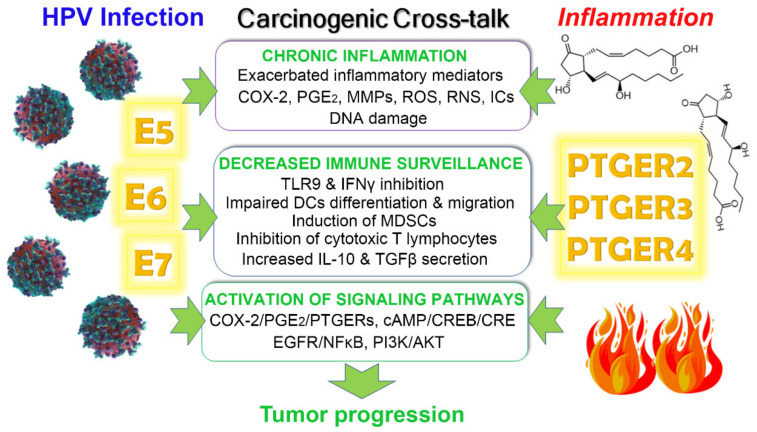
Chronic infection by high-risk human papillomaviruses (HPV) and chronic inflammation are factors associated with the onset and progression of several neoplasias, including cervical cancer. Oncogenic proteins E5, E6, and E7 from HPV are the main drivers of cervical carcinogenesis. In the present article, we review the general mechanisms of HPV-driven cervical carcinogenesis, as well as the involvement of cyclooxygenase-2 (COX-2)/prostaglandin E2 (PGE2) and downstream effectors in this pathology. We also review the evidence on the crosstalk between chronic HPV infection and PGE2 signaling, leading to immune response weakening and cervical cancer development. Finally, the last section updates the current therapeutic and preventive options targeting PGE2-derived inflammation and HPV infection in cervical cancer. These treatments include nonsteroidal anti-inflammatory drugs, prophylactic and therapeutical vaccines, immunomodulators, antivirals, and nanotechnology. Inflammatory signaling pathways are closely related to the carcinogenic nature of the virus, highlighting inflammation as a co-factor for HPV-dependent carcinogenesis. Therefore, blocking inflammatory signaling pathways, modulating immune response against HPV, and targeting the virus represent excellent options for anti-tumoral therapies in cervical cancer.
Keywords: cervical cancer; cervical cancer treatment; chronic inflammation; cyclooxygenase-2; human papillomavirus; oncogenic proteins; prostaglandin E2.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Association of human papillomavirus infection and inflammation in cervical cancer.Pathog Dis. 2019 Jul 1;77(5):ftz048. doi: 10.1093/femspd/ftz048. Pathog Dis. 2019. PMID: 31504464 Review.
-
The Autophagy Process in Cervical Carcinogenesis: Role of Non-Coding-RNAs, Molecular Mechanisms, and Therapeutic Targets.Cells. 2022 Apr 13;11(8):1323. doi: 10.3390/cells11081323. Cells. 2022. PMID: 35456001 Free PMC article. Review.
-
Mechanistic role of HPV-associated early proteins in cervical cancer: Molecular pathways and targeted therapeutic strategies.Crit Rev Oncol Hematol. 2022 Jun;174:103675. doi: 10.1016/j.critrevonc.2022.103675. Epub 2022 Apr 4. Crit Rev Oncol Hematol. 2022. PMID: 35381343 Review.
-
Human papillomavirus E5 protein induces expression of the EP4 subtype of prostaglandin E2 receptor in cyclic AMP response element-dependent pathways in cervical cancer cells.Carcinogenesis. 2009 Jan;30(1):141-9. doi: 10.1093/carcin/bgn236. Epub 2008 Oct 9. Carcinogenesis. 2009. PMID: 18849297
-
[Molecular basis of cervical carcinogenesis by high-risk human papillomaviruses].Uirusu. 2008 Dec;58(2):141-54. doi: 10.2222/jsv.58.141. Uirusu. 2008. PMID: 19374192 Review. Japanese.
Cited by
-
O-GlcNAcylation: Crosstalk between Hemostasis, Inflammation, and Cancer.Int J Mol Sci. 2024 Sep 13;25(18):9896. doi: 10.3390/ijms25189896. Int J Mol Sci. 2024. PMID: 39337387 Free PMC article. Review.
-
The Phytochemical α-Mangostin Inhibits Cervical Cancer Cell Proliferation and Tumor Growth by Downregulating E6/E7-HPV Oncogenes and KCNH1 Gene Expression.Int J Mol Sci. 2023 Feb 3;24(3):3055. doi: 10.3390/ijms24033055. Int J Mol Sci. 2023. PMID: 36769377 Free PMC article.
-
Viral oncogenesis in cancer: from mechanisms to therapeutics.Signal Transduct Target Ther. 2025 May 12;10(1):151. doi: 10.1038/s41392-025-02197-9. Signal Transduct Target Ther. 2025. PMID: 40350456 Free PMC article. Review.
-
A Case of Genital and Extragenital Warts Unresponsive to Immunotherapy Using Measles, Mumps, Rubella Vaccine.Int Med Case Rep J. 2023 Nov 15;16:739-746. doi: 10.2147/IMCRJ.S426665. eCollection 2023. Int Med Case Rep J. 2023. PMID: 38020581 Free PMC article.
-
The Preventive Role of the Vitamin D Endocrine System in Cervical Cancer.Int J Mol Sci. 2023 May 12;24(10):8665. doi: 10.3390/ijms24108665. Int J Mol Sci. 2023. PMID: 37240017 Free PMC article. Review.
