

Pathogenetic Mechanisms Underlying Spinocerebellar Ataxia Type 3 Are Altered in Primary Oligodendrocyte Culture
- PMID: 36010688
- PMCID: PMC9406561
- DOI: 10.3390/cells11162615
Pathogenetic Mechanisms Underlying Spinocerebellar Ataxia Type 3 Are Altered in Primary Oligodendrocyte Culture
Abstract
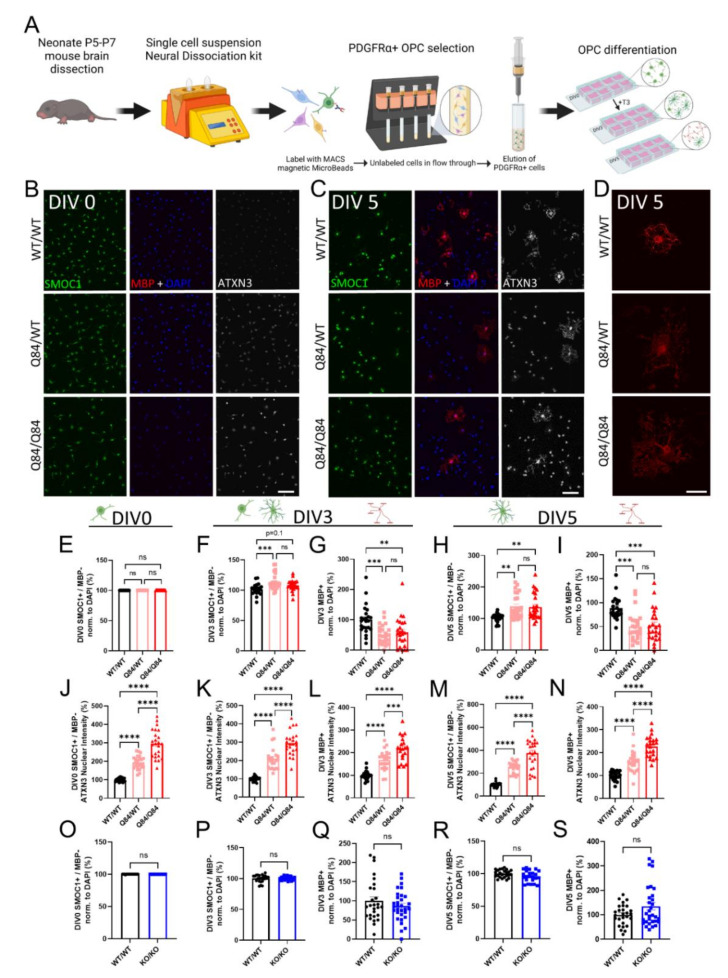
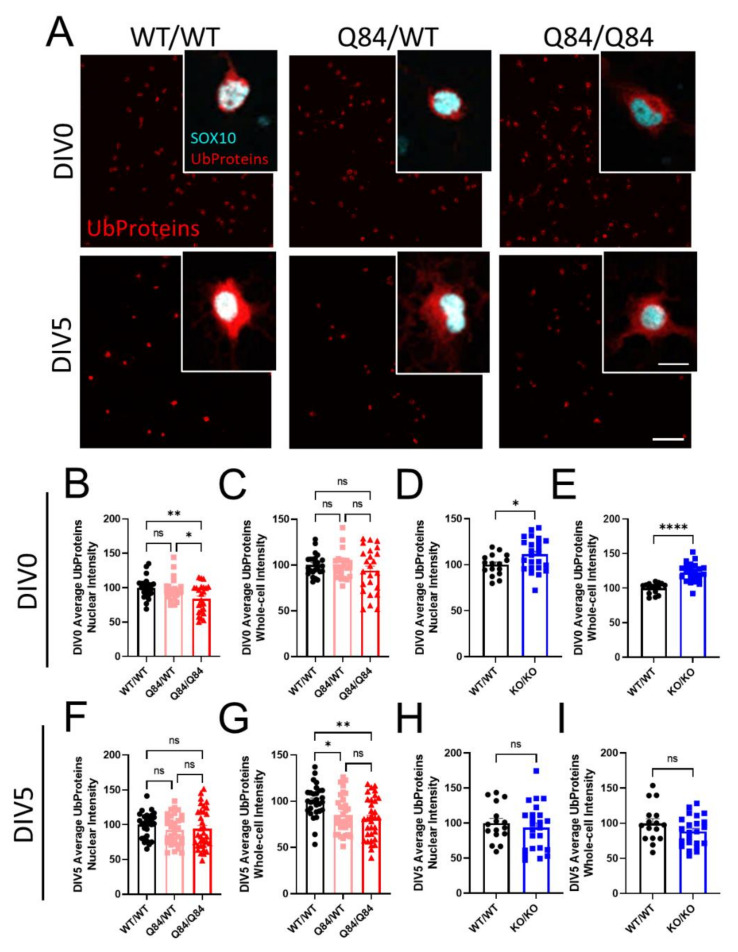
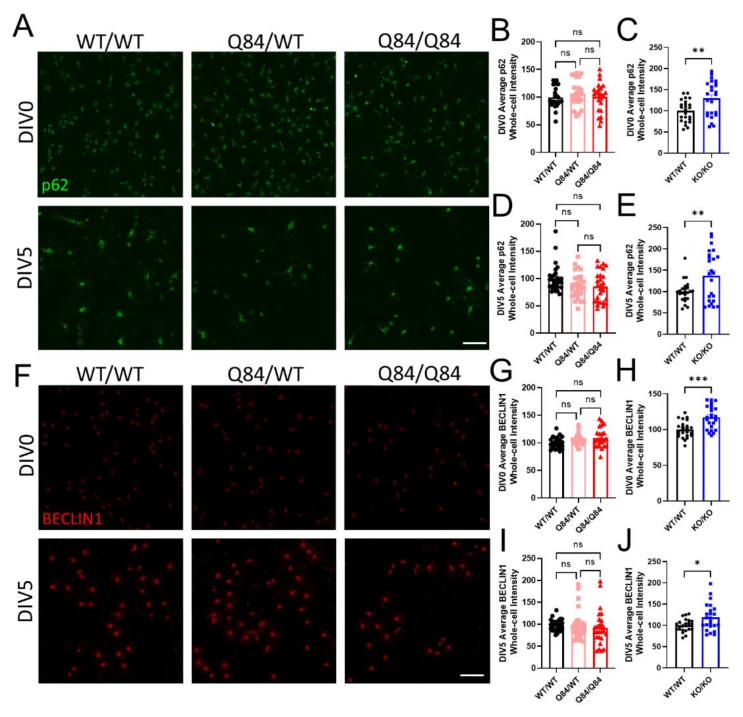
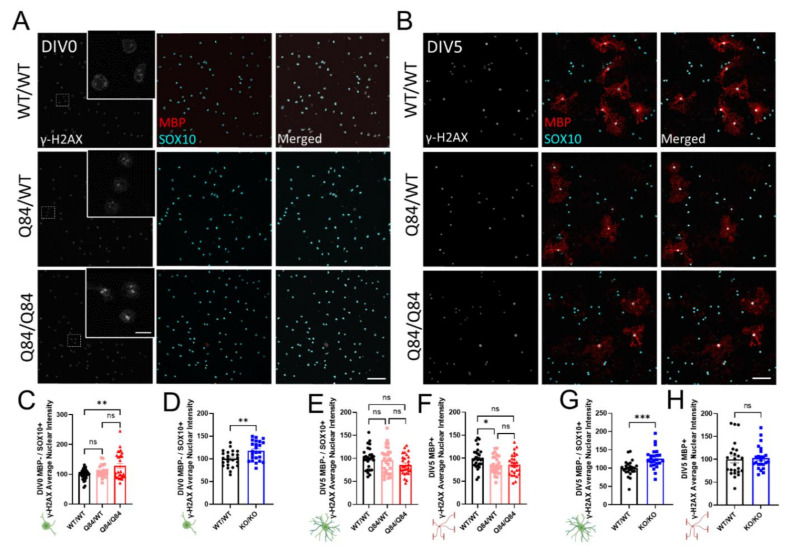
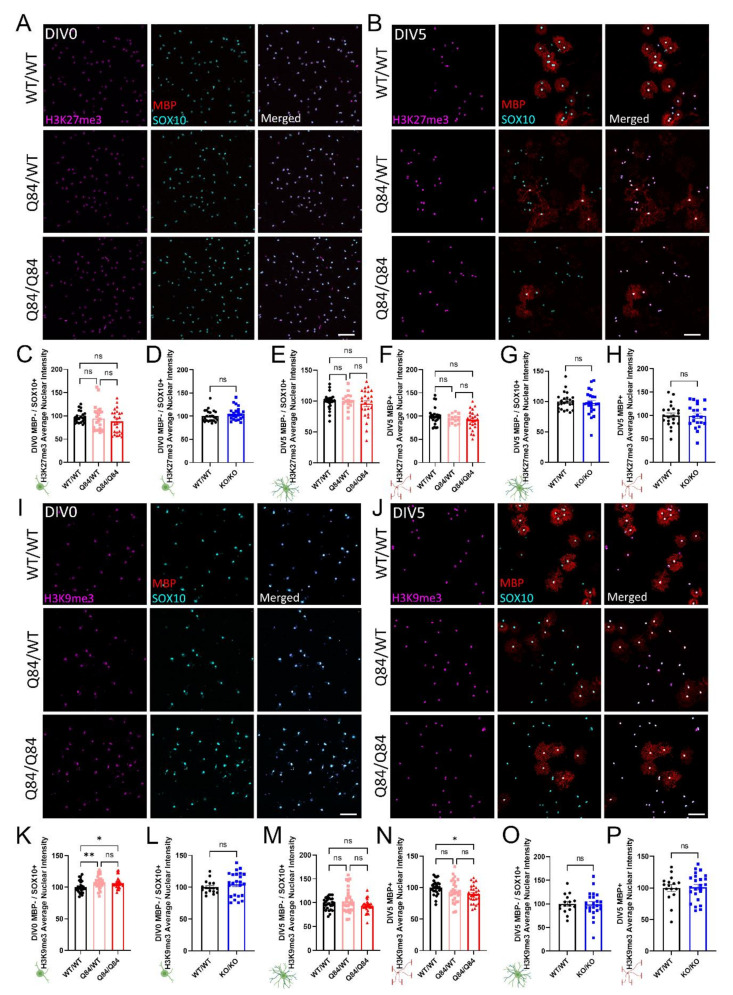
Emerging evidence has implicated non-neuronal cells, particularly oligodendrocytes, in the pathophysiology of many neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, Huntington's disease and Spinocerebellar ataxia type 3 (SCA3). We recently demonstrated that cell-autonomous dysfunction of oligodendrocyte maturation is one of the of the earliest and most robust changes in vulnerable regions of the SCA3 mouse brain. However, the cell- and disease-specific mechanisms that underlie oligodendrocyte dysfunction remain poorly understood and are difficult to isolate in vivo. In this study, we used primary oligodendrocyte cultures to determine how known pathogenic SCA3 mechanisms affect this cell type. We isolated oligodendrocyte progenitor cells from 5- to 7-day-old mice that overexpress human mutant ATXN3 or lack mouse ATXN3 and differentiated them for up to 5 days in vitro. Utilizing immunocytochemistry, we characterized the contributions of ATXN3 toxic gain-of-function and loss-of-function in oligodendrocyte maturation, protein quality pathways, DNA damage signaling, and methylation status. We illustrate the utility of primary oligodendrocyte culture for elucidating cell-specific pathway dysregulation relevant to SCA3. Given recent work demonstrating disease-associated oligodendrocyte signatures in other neurodegenerative diseases, this novel model has broad applicability in revealing mechanistic insights of oligodendrocyte contribution to pathogenesis.
Keywords: Machado–Joseph disease; ataxia; myelination; oligodendrocyte; oligodendrocyte precursor cells; polyglutamine; spinocerebellar ataxia type 3.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
ASOs are an effective treatment for disease-associated oligodendrocyte signatures in premanifest and symptomatic SCA3 mice.Mol Ther. 2024 May 1;32(5):1359-1372. doi: 10.1016/j.ymthe.2024.02.033. Epub 2024 Feb 29. Mol Ther. 2024. PMID: 38429929 Free PMC article.
-
Impaired Oligodendrocyte Maturation Is an Early Feature in SCA3 Disease Pathogenesis.J Neurosci. 2022 Feb 23;42(8):1604-1617. doi: 10.1523/JNEUROSCI.1954-20.2021. Epub 2022 Jan 18. J Neurosci. 2022. PMID: 35042771 Free PMC article.
-
Comparison of spinocerebellar ataxia type 3 mouse models identifies early gain-of-function, cell-autonomous transcriptional changes in oligodendrocytes.Hum Mol Genet. 2017 Sep 1;26(17):3362-3374. doi: 10.1093/hmg/ddx224. Hum Mol Genet. 2017. PMID: 28854700 Free PMC article.
-
Autophagy in Spinocerebellar Ataxia Type 3: From Pathogenesis to Therapeutics.Int J Mol Sci. 2023 Apr 17;24(8):7405. doi: 10.3390/ijms24087405. Int J Mol Sci. 2023. PMID: 37108570 Free PMC article. Review.
-
ATXN3: a multifunctional protein involved in the polyglutamine disease spinocerebellar ataxia type 3.Expert Rev Mol Med. 2024 Sep 25;26:e19. doi: 10.1017/erm.2024.10. Expert Rev Mol Med. 2024. PMID: 39320846 Free PMC article. Review.
Cited by
-
ASOs are an effective treatment for disease-associated oligodendrocyte signatures in premanifest and symptomatic SCA3 mice.Mol Ther. 2024 May 1;32(5):1359-1372. doi: 10.1016/j.ymthe.2024.02.033. Epub 2024 Feb 29. Mol Ther. 2024. PMID: 38429929 Free PMC article.
-
FGF9 is required for Purkinje cell development and function in the cerebellum.iScience. 2024 Jan 26;27(2):109039. doi: 10.1016/j.isci.2024.109039. eCollection 2024 Feb 16. iScience. 2024. PMID: 38352230 Free PMC article.
-
Disease-associated oligodendrocyte signatures are spatiotemporally dysregulated in spinocerebellar ataxia type 3.Front Neurosci. 2023 Feb 15;17:1118429. doi: 10.3389/fnins.2023.1118429. eCollection 2023. Front Neurosci. 2023. PMID: 36875652 Free PMC article.
-
Antisense Oligonucleotide Silencing Reverses Abnormal Neurochemistry in Spinocerebellar Ataxia 3 Mice.Ann Neurol. 2023 Oct;94(4):658-671. doi: 10.1002/ana.26713. Epub 2023 Aug 2. Ann Neurol. 2023. PMID: 37243335 Free PMC article.
-
Myelinating Glia: Potential Therapeutic Targets in Polyglutamine Spinocerebellar Ataxias.Cells. 2023 Feb 13;12(4):601. doi: 10.3390/cells12040601. Cells. 2023. PMID: 36831268 Free PMC article. Review.
References
-
- Rüb U., Schöls L., Paulson H., Auburger G., Kermer P., Jen J.C., Seidel K., Korf H., Deller T. Clinical features, neurogenetics and neuropathology of the polyglutamine spinocerebellar ataxias type 1, 2, 3, 6 and 7. Prog. Neurobiol. 2013;104:38–66. doi: 10.1016/j.pneurobio.2013.01.001. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
