

A Study on the Pathogenesis of Vascular Cognitive Impairment and Dementia: The Chronic Cerebral Hypoperfusion Hypothesis
- PMID: 36012981
- PMCID: PMC9409771
- DOI: 10.3390/jcm11164742
A Study on the Pathogenesis of Vascular Cognitive Impairment and Dementia: The Chronic Cerebral Hypoperfusion Hypothesis
Abstract
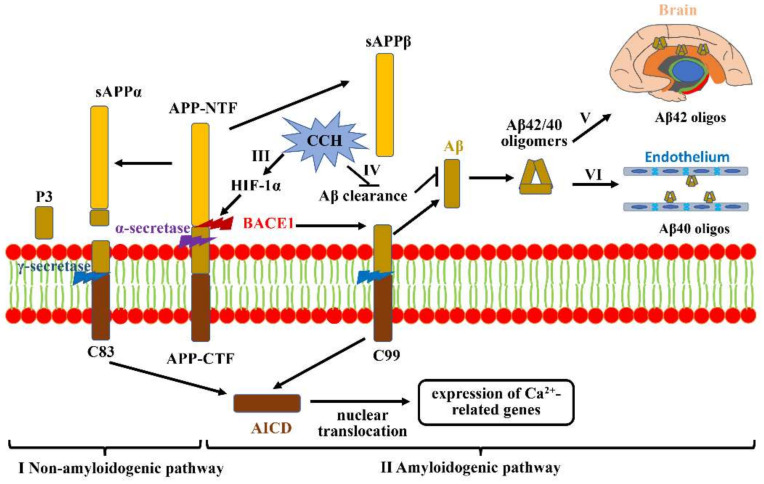
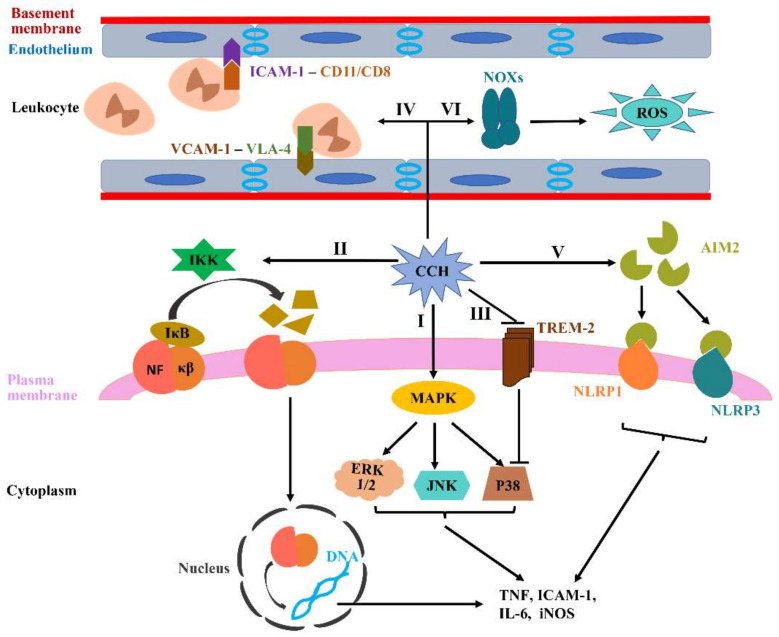
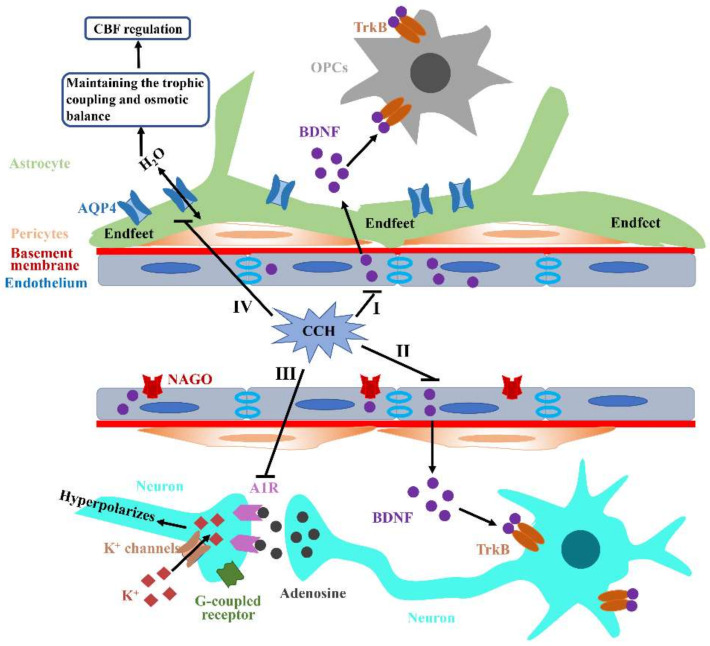
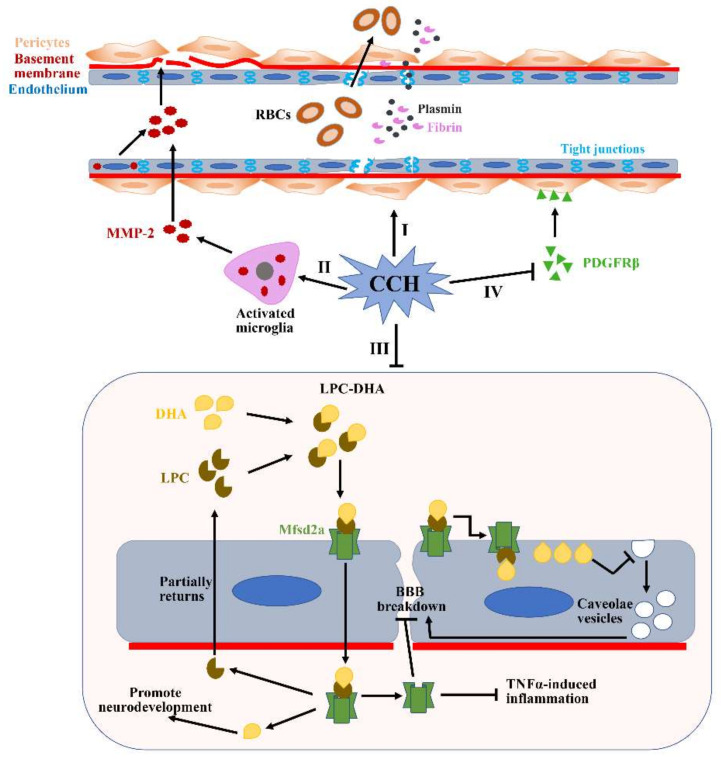
The pathogenic mechanisms underlying vascular cognitive impairment and dementia (VCID) remain controversial due to the heterogeneity of vascular causes and complexity of disease neuropathology. However, one common feature shared among all these vascular causes is cerebral blood flow (CBF) dysregulation, and chronic cerebral hypoperfusion (CCH) is the universal consequence of CBF dysregulation, which subsequently results in an insufficient blood supply to the brain, ultimately contributing to VCID. The purpose of this comprehensive review is to emphasize the important contributions of CCH to VCID and illustrate the current findings about the mechanisms involved in CCH-induced VCID pathological changes. Specifically, evidence is mainly provided to support the molecular mechanisms, including Aβ accumulation, inflammation, oxidative stress, blood-brain barrier (BBB) disruption, trophic uncoupling and white matter lesions (WMLs). Notably, there are close interactions among these multiple mechanisms, and further research is necessary to elucidate the hitherto unsolved questions regarding these interactions. An enhanced understanding of the pathological features in preclinical models could provide a theoretical basis, ultimately achieving the shift from treatment to prevention.
Keywords: blood-brain barrier; cerebral blood flow regulation; chronic cerebral hypoperfusion; neuroinflammation; oxidative stress; trophic uncoupling; vascular cognitive impairment and dementia; white matter lesions; β-amyloid.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
