

Frataxin controls ketone body metabolism through regulation of OXCT1
- PMID: 36016708
- PMCID: PMC9396447
- DOI: 10.1093/pnasnexus/pgac142
Frataxin controls ketone body metabolism through regulation of OXCT1
Abstract
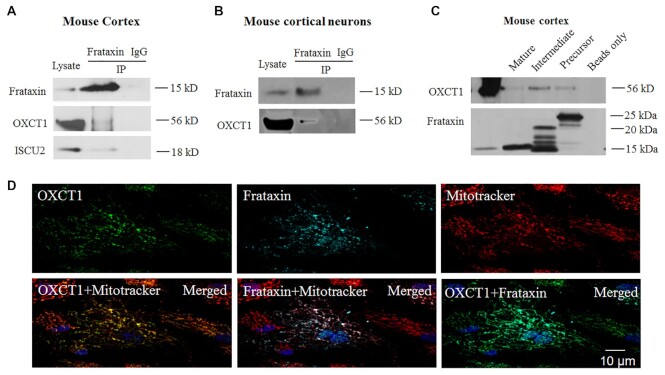
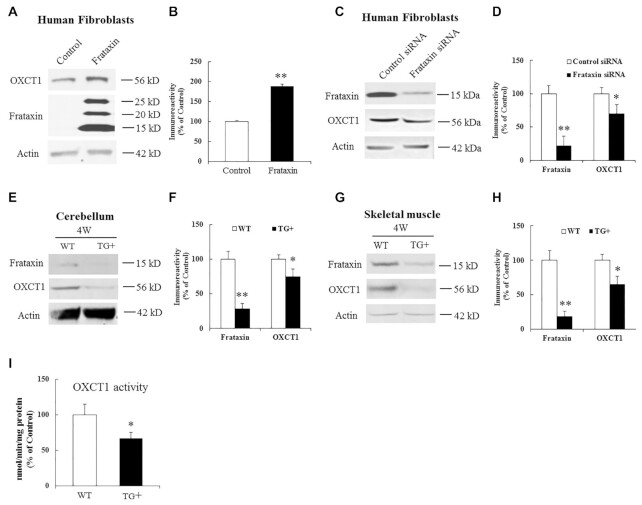
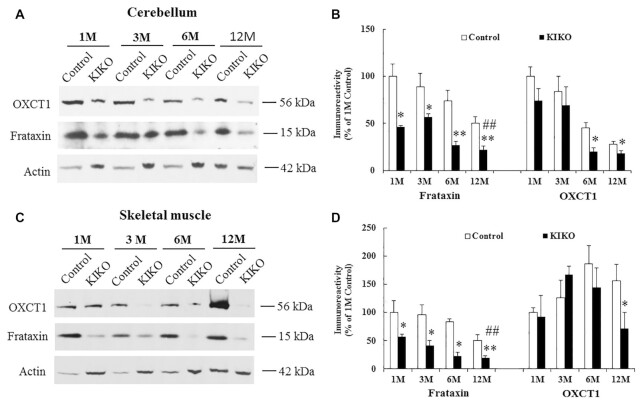
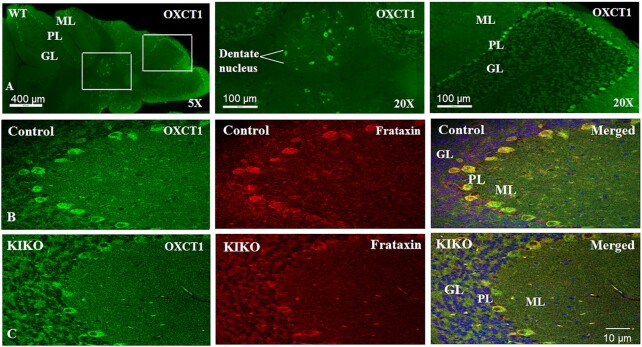
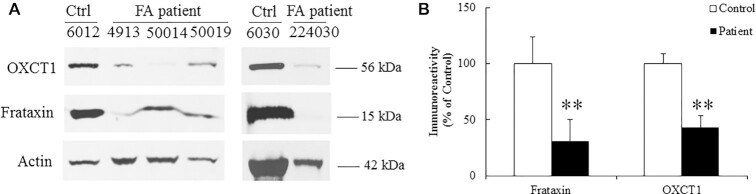
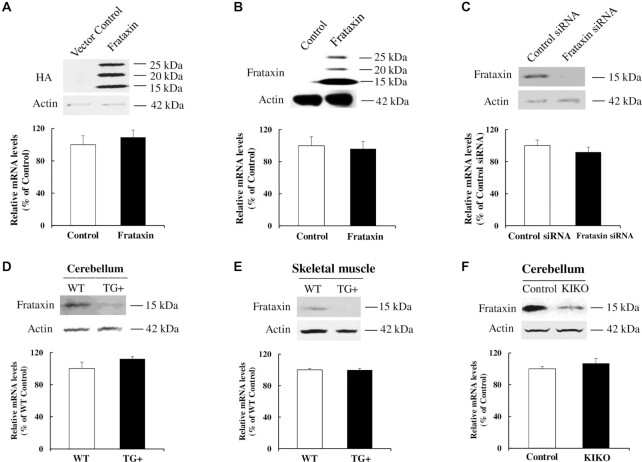
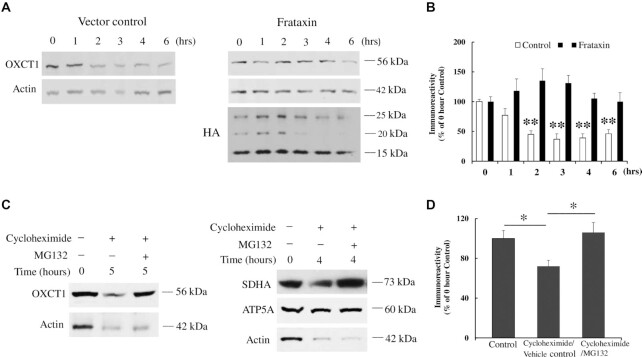
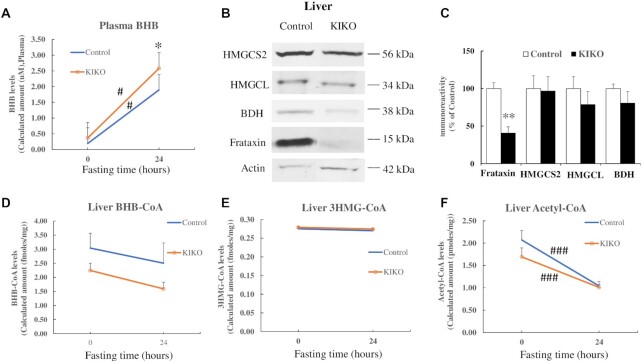
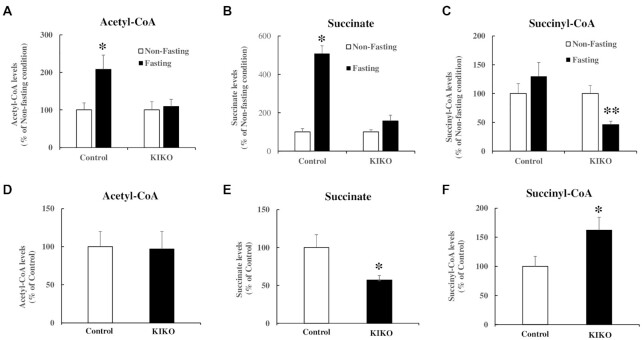
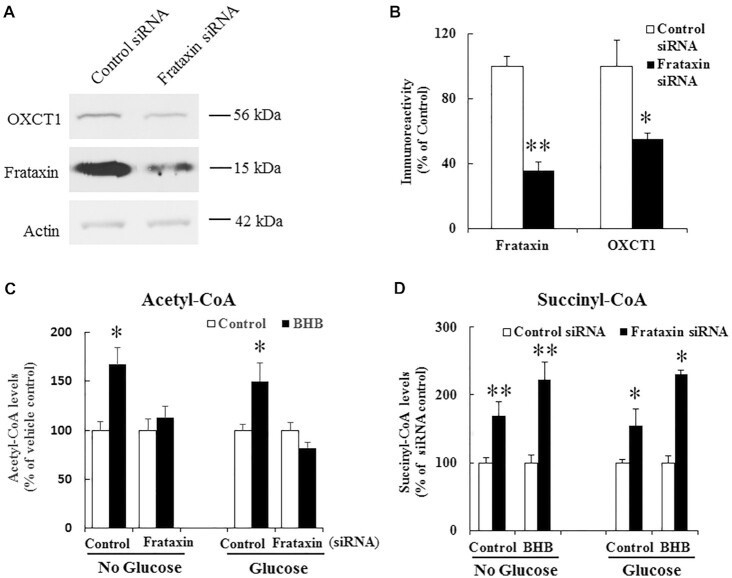
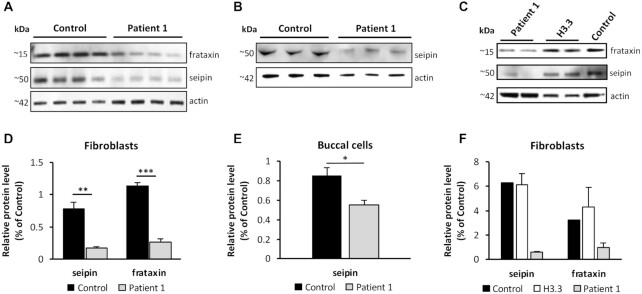
Friedreich's ataxia (FRDA) is an autosomal recessive neurodegenerative disease caused by the deficiency of mitochondrial protein frataxin, which plays a crucial role in iron-sulphur cluster formation and ATP production. The cellular function of frataxin is not entirely known. Here, we demonstrate that frataxin controls ketone body metabolism through regulation of 3-Oxoacid CoA-Transferase 1 (OXCT1), a rate limiting enzyme catalyzing the conversion of ketone bodies to acetoacetyl-CoA that is then fed into the Krebs cycle. Biochemical studies show a physical interaction between frataxin and OXCT1 both in vivo and in vitro. Frataxin overexpression also increases OXCT1 protein levels in human skin fibroblasts while frataxin deficiency decreases OXCT1 in multiple cell types including cerebellum and skeletal muscle both acutely and chronically, suggesting that frataxin directly regulates OXCT1. This regulation is mediated by frataxin-dependent suppression of ubiquitin-proteasome system (UPS)-dependent OXCT1 degradation. Concomitantly, plasma ketone bodies are significantly elevated in frataxin deficient knock-in/knockout (KIKO) mice with no change in the levels of other enzymes involved in ketone body production. In addition, ketone bodies fail to be metabolized to acetyl-CoA accompanied by increased succinyl-CoA in vitro in frataxin deficient cells, suggesting that ketone body elevation is caused by frataxin-dependent reduction of OXCT1 leading to deficits in tissue utilization of ketone bodies. Considering the potential role of metabolic abnormalities and deficiency of ATP production in FRDA, our results suggest a new role for frataxin in ketone body metabolism and also suggest modulation of OXCT1 may be a potential therapeutic approach for FRDA.
Keywords: Friedreich's ataxia; OXCT1; frataxin; ketone body.
© The Author(s) 2022. Published by Oxford University Press on behalf of the National Academy of Sciences.
Figures
References
-
- Campuzano V, et al. 1996. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 271:1423–1427. - PubMed
-
- Cossée M, et al. 1999. Friedreich's ataxia: point mutations and clinical presentation of compound heterozygotes. Ann Neurol. 45:200–206. - PubMed
-
- Rötig A, et al. 1997. Aconitase and mitochondrial iron–sulphur protein deficiency in Friedreich ataxia. Nat Genet. 17:215–217. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
