

Cigarette Smoke Modulates Inflammation and Immunity via Reactive Oxygen Species-Regulated Trained Immunity and Trained Tolerance Mechanisms
- PMID: 36017612
- PMCID: PMC10171958
- DOI: 10.1089/ars.2022.0087
Cigarette Smoke Modulates Inflammation and Immunity via Reactive Oxygen Species-Regulated Trained Immunity and Trained Tolerance Mechanisms
Abstract
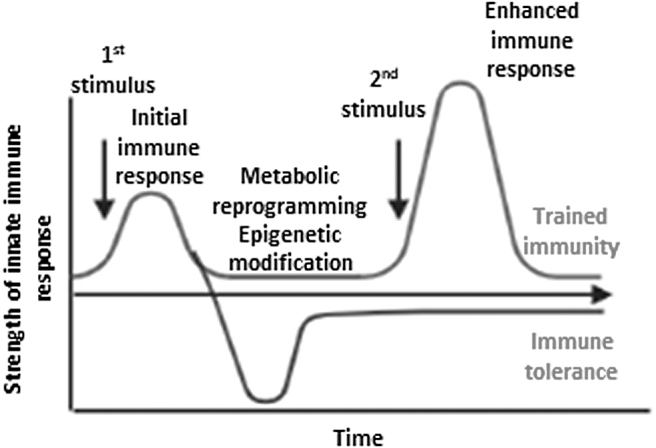
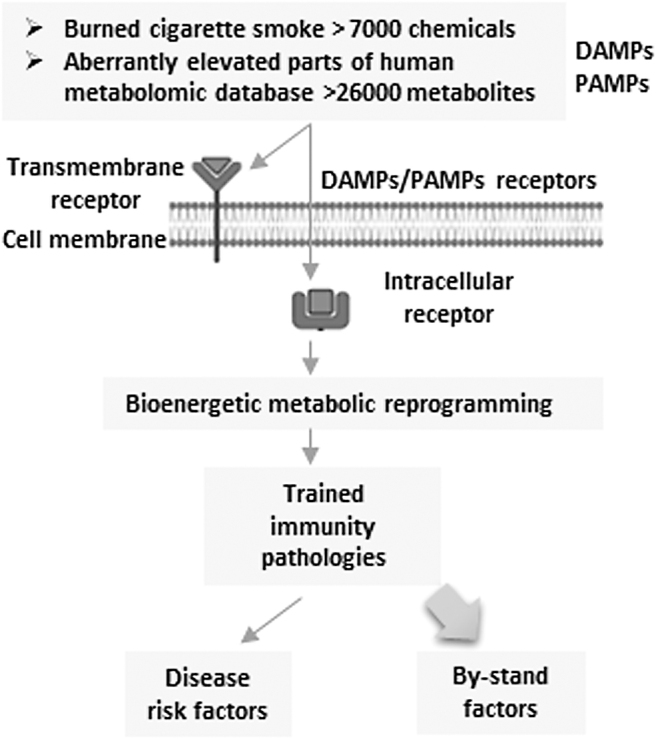
Significance: Cigarette smoke (CS) is a prominent cause of morbidity and death and poses a serious challenge to the current health care system worldwide. Its multifaceted roles have led to cardiovascular, respiratory, immunological, and neoplastic diseases. Recent Advances: CS influences both innate and adaptive immunity and regulates immune responses by exacerbating pathogenic immunological responses and/or suppressing defense immunity. There is substantial evidence pointing toward a critical role of CS in vascular immunopathology, but a comprehensive and up-to-date review is lacking. Critical Issues: This review aims to synthesize novel conceptual advances on the immunomodulatory action of CS with a focus on the cardiovascular system from the following perspectives: (i) the signaling of danger-associated molecular pattern (DAMP) receptors contributes to CS modulation of inflammation and immunity; (ii) CS reprograms immunometabolism and trained immunity-related metabolic pathways in innate immune cells and T cells, which can be sensed by the cytoplasmic (cytosolic and non-nuclear organelles) reactive oxygen species (ROS) system in vascular cells; (iii) how nuclear ROS drive CS-promoted DNA damage and cell death pathways, thereby amplifying inflammation and immune responses; and (iv) CS induces endothelial cell (EC) dysfunction and vascular inflammation to promote cardiovascular diseases (CVDs). Future Directions: Despite significant progress in understanding the cellular and molecular mechanisms linking CS to immunity, further investigations are warranted to elucidate novel mechanisms responsible for CS-mediated immunopathology of CVDs; in particular, the research in redox regulation of immune functions of ECs and their fate affected by CS is still in its infancy.
Keywords: cell death; cigarette smoke; immunometabolism; morphine; trained immunity; trained tolerance.
Conflict of interest statement
No competing financial interests exist.
Figures




Similar articles
-
Impacts of cigarette smoking on immune responsiveness: Up and down or upside down?Oncotarget. 2017 Jan 3;8(1):268-284. doi: 10.18632/oncotarget.13613. Oncotarget. 2017. PMID: 27902485 Free PMC article. Review.
-
Effects of Cigarette Smoking on Transplant Survival: Extending or Shortening It?Front Immunol. 2017 Feb 10;8:127. doi: 10.3389/fimmu.2017.00127. eCollection 2017. Front Immunol. 2017. PMID: 28239383 Free PMC article.
-
Macrophage Rac2 Is Required to Reduce the Severity of Cigarette Smoke-induced Pneumonia.Am J Respir Crit Care Med. 2018 Nov 15;198(10):1288-1301. doi: 10.1164/rccm.201712-2388OC. Am J Respir Crit Care Med. 2018. PMID: 29897791 Free PMC article.
-
Cigarette smoke-induced damage-associated molecular pattern release from necrotic neutrophils triggers proinflammatory mediator release.Am J Respir Cell Mol Biol. 2015 May;52(5):554-62. doi: 10.1165/rcmb.2013-0505OC. Am J Respir Cell Mol Biol. 2015. PMID: 25192219 Clinical Trial.
-
Trained Immunity and Reactivity of Macrophages and Endothelial Cells.Arterioscler Thromb Vasc Biol. 2021 Mar;41(3):1032-1046. doi: 10.1161/ATVBAHA.120.315452. Epub 2020 Dec 31. Arterioscler Thromb Vasc Biol. 2021. PMID: 33380171 Free PMC article. Review.
Cited by
-
Exploring the Causal Relationship Between Modifiable Exposures and Diabetes Mellitus: A Two-Sample Mendelian Randomization Analysis.Cureus. 2024 Apr 25;16(4):e59034. doi: 10.7759/cureus.59034. eCollection 2024 Apr. Cureus. 2024. PMID: 38800249 Free PMC article.
-
Chronic Kidney Disease Transdifferentiates Veins into a Specialized Immune-Endocrine Organ with Increased MYCN-AP1 Signaling.Cells. 2023 May 26;12(11):1482. doi: 10.3390/cells12111482. Cells. 2023. PMID: 37296603 Free PMC article.
-
Oral Submucous Fibrosis: Etiological Mechanism, Malignant Transformation, Therapeutic Approaches and Targets.Int J Mol Sci. 2023 Mar 5;24(5):4992. doi: 10.3390/ijms24054992. Int J Mol Sci. 2023. PMID: 36902423 Free PMC article. Review.
-
Trained immunity in chronic inflammatory diseases and cancer.Nat Rev Immunol. 2025 Jul;25(7):497-514. doi: 10.1038/s41577-025-01132-x. Epub 2025 Jan 31. Nat Rev Immunol. 2025. PMID: 39891000 Review.
-
Editorial: Insights in cardiovascular therapeutics 2022-cardiovascular innate immunity.Front Cardiovasc Med. 2023 Apr 18;10:1184030. doi: 10.3389/fcvm.2023.1184030. eCollection 2023. Front Cardiovasc Med. 2023. PMID: 37144060 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
