

PRC1-independent binding and activity of RYBP on the KSHV genome during de novo infection
- PMID: 36026503
- PMCID: PMC9455864
- DOI: 10.1371/journal.ppat.1010801
PRC1-independent binding and activity of RYBP on the KSHV genome during de novo infection
Abstract
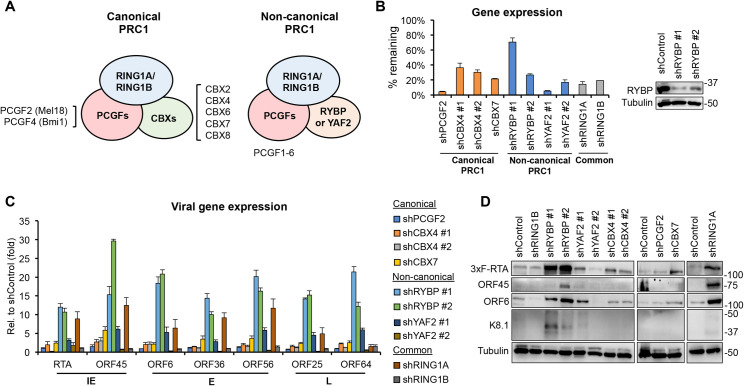
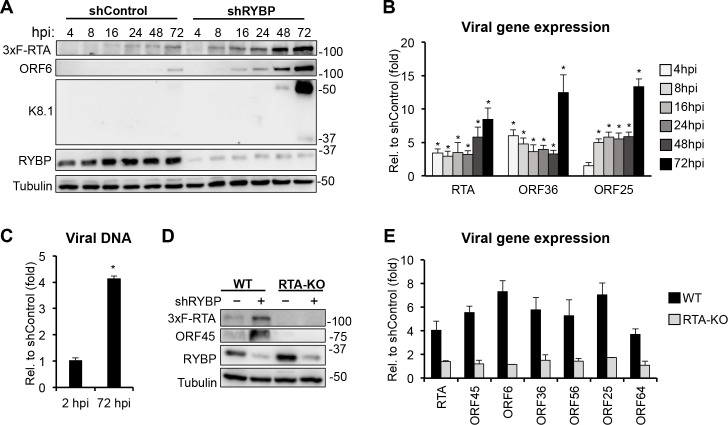
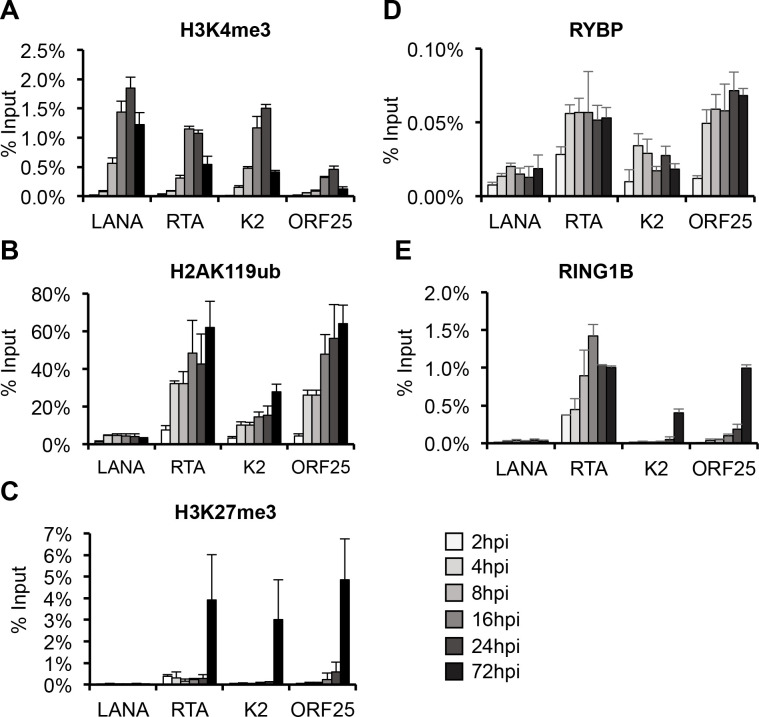
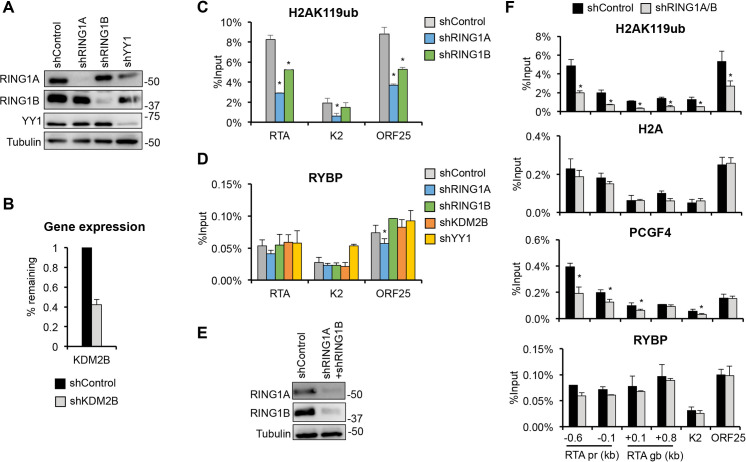
Kaposi's sarcoma-associated herpesvirus (KSHV) is an oncogenic virus that causes lifelong infection in humans by establishing latency after primary infection. Latent infection is a prerequisite for both persistent infection and the development of KSHV-associated cancers. While viral lytic genes are transiently expressed after primary infection, their expression is significantly restricted and concomitant with the binding of host epigenetic repressors Polycomb Repressive Complex 1 and 2 (PRC1 and PRC2) to lytic genes. PRC1 and PRC2 mediate the repressive histone marks H2AK119ub and H3K27me3, respectively, and maintain heterochromatin structure on KSHV lytic genes to inhibit their expression. In contrast to PRC2, little is known about the recruitment and role of PRC1 factors on the KSHV genome following de novo infection. Thus, the goal of this study was to examine the function of PRC1 factors in the establishment of KSHV latency. To address this question, we performed an shRNA screen targeting 7 different components of the canonical and non-canonical PRC1 complexes during primary KSHV infection. We found that RYBP, a main subunit of the non-canonical PRC1 complexes, is a potent repressor of KSHV lytic genes that can bind to the viral genome and inhibit lytic genes as early as 4 hours post infection. Surprisingly, our ChIP analyses showed that RYBP binds to lytic viral gene promoters in a PRC1-independent manner, does not affect PRC1 activity on the KSHV genome, and can reduce the level of histone marks associated with transcription elongation. Our data also suggest that RYBP can repress the viral lytic cycle after primary infection by inhibiting the transcription elongation of the lytic cycle inducer KSHV gene RTA. Based on our results we propose that RYBP uses a PRC1-independent mechanism to block KSHV RTA expression thereby promoting the establishment of KSHV latency following de novo infection.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
LANA-Mediated Recruitment of Host Polycomb Repressive Complexes onto the KSHV Genome during De Novo Infection.PLoS Pathog. 2016 Sep 8;12(9):e1005878. doi: 10.1371/journal.ppat.1005878. eCollection 2016 Sep. PLoS Pathog. 2016. PMID: 27606464 Free PMC article.
-
Biphasic euchromatin-to-heterochromatin transition on the KSHV genome following de novo infection.PLoS Pathog. 2013;9(12):e1003813. doi: 10.1371/journal.ppat.1003813. Epub 2013 Dec 19. PLoS Pathog. 2013. PMID: 24367262 Free PMC article.
-
Epigenetic factor siRNA screen during primary KSHV infection identifies novel host restriction factors for the lytic cycle of KSHV.PLoS Pathog. 2020 Jan 10;16(1):e1008268. doi: 10.1371/journal.ppat.1008268. eCollection 2020 Jan. PLoS Pathog. 2020. PMID: 31923286 Free PMC article.
-
Lytic cycle switches of oncogenic human gammaherpesviruses.Adv Cancer Res. 2007;97:81-109. doi: 10.1016/S0065-230X(06)97004-3. Adv Cancer Res. 2007. PMID: 17419942 Review.
-
An atlas of chromatin landscape in KSHV-infected cells during de novo infection and reactivation.Virology. 2024 Sep;597:110146. doi: 10.1016/j.virol.2024.110146. Epub 2024 Jun 19. Virology. 2024. PMID: 38909515 Review.
Cited by
-
H2A monoubiquitination: insights from human genetics and animal models.Hum Genet. 2024 Apr;143(4):511-527. doi: 10.1007/s00439-023-02557-x. Epub 2023 Apr 22. Hum Genet. 2024. PMID: 37086328 Review.
-
Microneedle-Array-Mediated Transdermal Delivery of GCV-Functionalized Zeolitic Imidazolate Framework-8 Nanoparticles for KSHV Treatment.Int J Mol Sci. 2024 Dec 2;25(23):12946. doi: 10.3390/ijms252312946. Int J Mol Sci. 2024. PMID: 39684656 Free PMC article.
-
Hypoxia and HIF-1α promote lytic de novo KSHV infection.J Virol. 2023 Nov 30;97(11):e0097223. doi: 10.1128/jvi.00972-23. Epub 2023 Nov 1. J Virol. 2023. PMID: 37909728 Free PMC article.
-
Kaposi's Sarcoma-Associated Herpesvirus Immediate Early Proteins Trigger FOXQ1 Expression in Oral Epithelial Cells, Engaging in a Novel Lytic Cycle-Sustaining Positive Feedback Loop.J Virol. 2023 Mar 30;97(3):e0169622. doi: 10.1128/jvi.01696-22. Epub 2023 Feb 23. J Virol. 2023. PMID: 36815831 Free PMC article.
-
Kaposi's sarcoma-associated herpesvirus terminal repeat regulates inducible lytic gene promoters.J Virol. 2024 Feb 20;98(2):e0138623. doi: 10.1128/jvi.01386-23. Epub 2024 Jan 19. J Virol. 2024. PMID: 38240593 Free PMC article.
References
-
- Toth Z, Brulois K, Lee HR, Izumiya Y, Tepper C, Kung HJ, et al.. Biphasic euchromatin-to-heterochromatin transition on the KSHV genome following de novo infection. PLoS Pathog. 2013;9(12):e1003813. Epub 2013/12/25. doi: 10.1371/journal.ppat.1003813 ; PubMed Central PMCID: PMC3868514. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
