

Prion strains viewed through the lens of cryo-EM
- PMID: 36028585
- PMCID: PMC10113314
- DOI: 10.1007/s00441-022-03676-z
Prion strains viewed through the lens of cryo-EM
Abstract
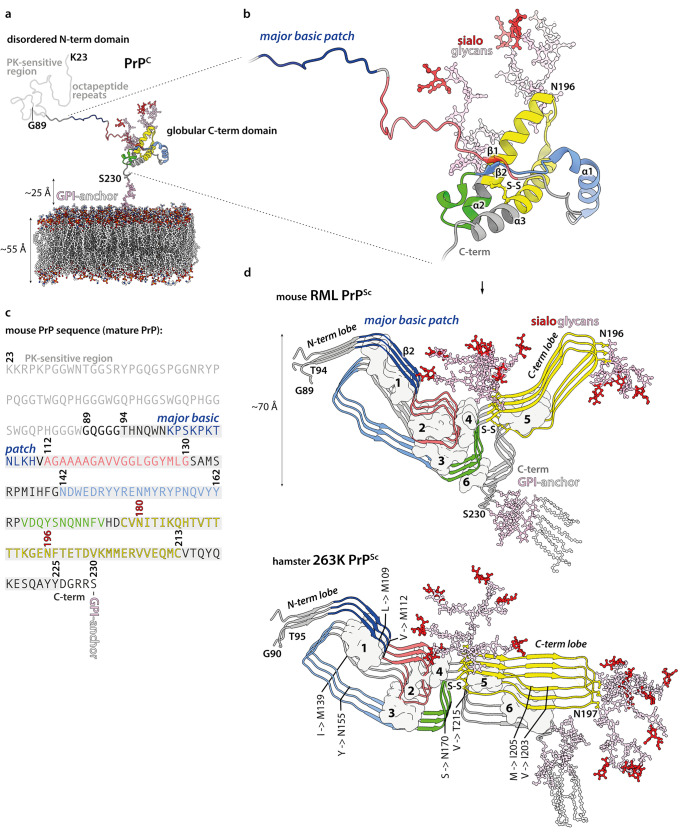
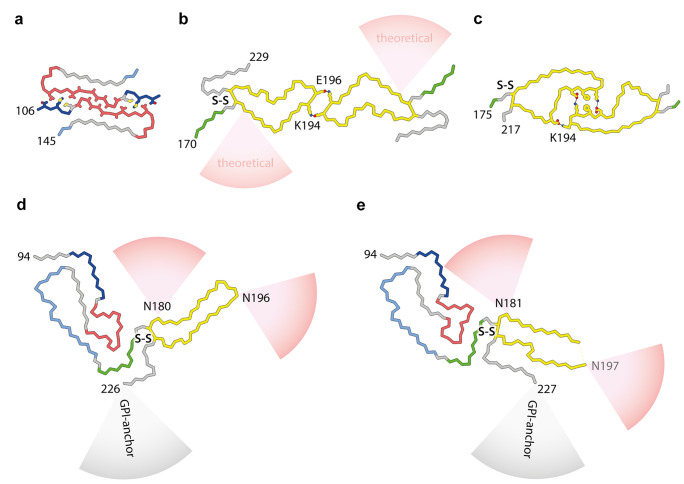
Mammalian prions are lethal transmissible pathogens that cause fatal neurodegenerative diseases in humans and animals. They consist of fibrils of misfolded, host-encoded prion protein (PrP) which propagate through templated protein polymerisation. Prion strains produce distinct clinicopathological phenotypes in the same host and appear to be encoded by distinct misfolded PrP conformations and assembly states. Despite fundamental advances in our understanding of prion biology, key knowledge gaps remain. These include precise delineation of prion replication mechanisms, detailed explanation of the molecular basis of prion strains and inter-species transmission barriers, and the structural definition of neurotoxic PrP species. Central to addressing these questions is the determination of prion structure. While high-resolution definition of ex vivo prion fibrils once seemed unlikely, recent advances in cryo-electron microscopy (cryo-EM) and computational methods for 3D reconstruction of amyloids have now made this possible. Recently, near-atomic resolution structures of highly infectious, ex vivo prion fibrils from hamster 263K and mouse RML prion strains were reported. The fibrils have a comparable parallel in-register intermolecular β-sheet (PIRIBS) architecture that now provides a structural foundation for understanding prion strain diversity in mammals. Here, we review these new findings and discuss directions for future research.
Keywords: Cryo-EM; Prion; Prion disease; Prion strains; Prion structure.
© 2022. The Author(s).
Conflict of interest statement
J. C. is a director and J. C. and J. D. F. W. are shareholders of D-Gen Limited, an academic spin-out company working in the field of prion disease diagnosis, decontamination, and therapeutics. S. W. M. and A. W. declare no competing interests.
Figures
References
-
- Asante EA, Gowland I, Grimshaw A, Linehan JM, Smidak M, Houghton R, Osiguwa O, Tomlinson A, Joiner S, Brandner S, Wadsworth JD, Collinge J. Absence of spontaneous disease and comparative prion susceptibility of transgenic mice expressing mutant human prion proteins. J Gen Virol. 2009;90:546–558. doi: 10.1099/vir.0.007930-0. - DOI - PMC - PubMed
-
- Asante EA, Grimshaw A, Smidak M, Jakubcova T, Tomlinson A, Jeelani A, Hamdan S, Powell C, Joiner S, Linehan JM, Brandner S, Wadsworth JD, Collinge J. Transmission properties of human PrP 102L prions challenge the relevance of mouse models of GSS. PLoS Pathog. 2015;11:e1004953. doi: 10.1371/journal.ppat.1004953. - DOI - PMC - PubMed
-
- Asante EA, Linehan J, Desbruslais M, Joiner S, Gowland I, Wood A, Welch J, Hill AF, Lloyd S, Wadsworth JD, Collinge J. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J. 2002;21:6358–6366. doi: 10.1093/emboj/cdf653. - DOI - PMC - PubMed
-
- Asante EA, Linehan JM, Smidak M, Tomlinson A, Grimshaw A, Jeelani A, Jakubcova T, Hamdan S, Powell C, Brandner S, Wadsworth JD, Collinge J. Inherited prion disease A117V is not simply a proteinopathy but produces prions transmissible to transgenic mice expressing homologous prion protein. PLoS Pathog. 2013;9:e1003643. doi: 10.1371/journal.ppat.1003643. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
