

MOG analogues to explore the MCT2 pharmacophore, α-ketoglutarate biology and cellular effects of N-oxalylglycine
- PMID: 36028752
- PMCID: PMC9418262
- DOI: 10.1038/s42003-022-03805-y
MOG analogues to explore the MCT2 pharmacophore, α-ketoglutarate biology and cellular effects of N-oxalylglycine
Erratum in
-
Publisher Correction: MOG analogues to explore the MCT2 pharmacophore, α-ketoglutarate biology and cellular effects of N-oxalylglycine.Commun Biol. 2022 Sep 27;5(1):1019. doi: 10.1038/s42003-022-03922-8. Commun Biol. 2022. PMID: 36167803 Free PMC article. No abstract available.
Abstract
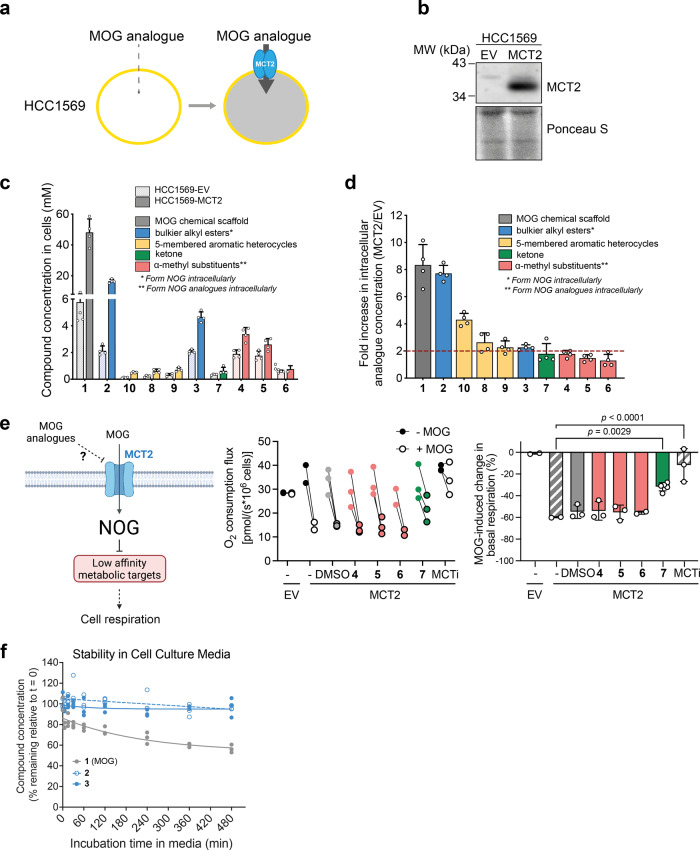
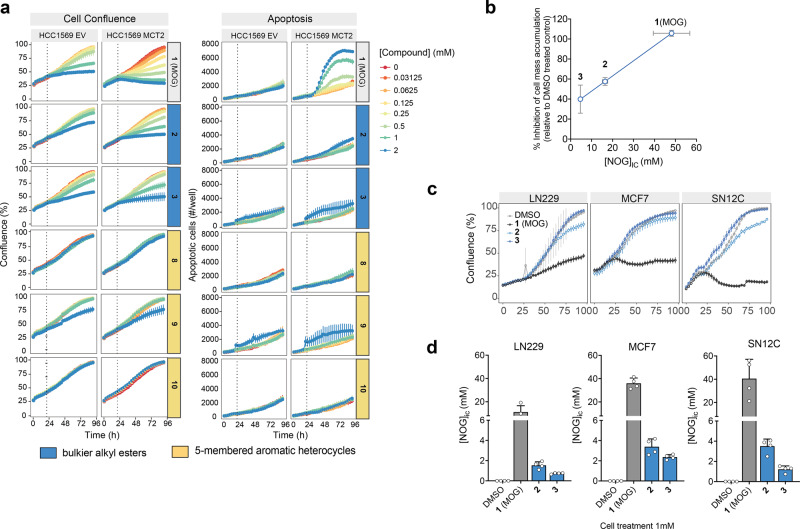
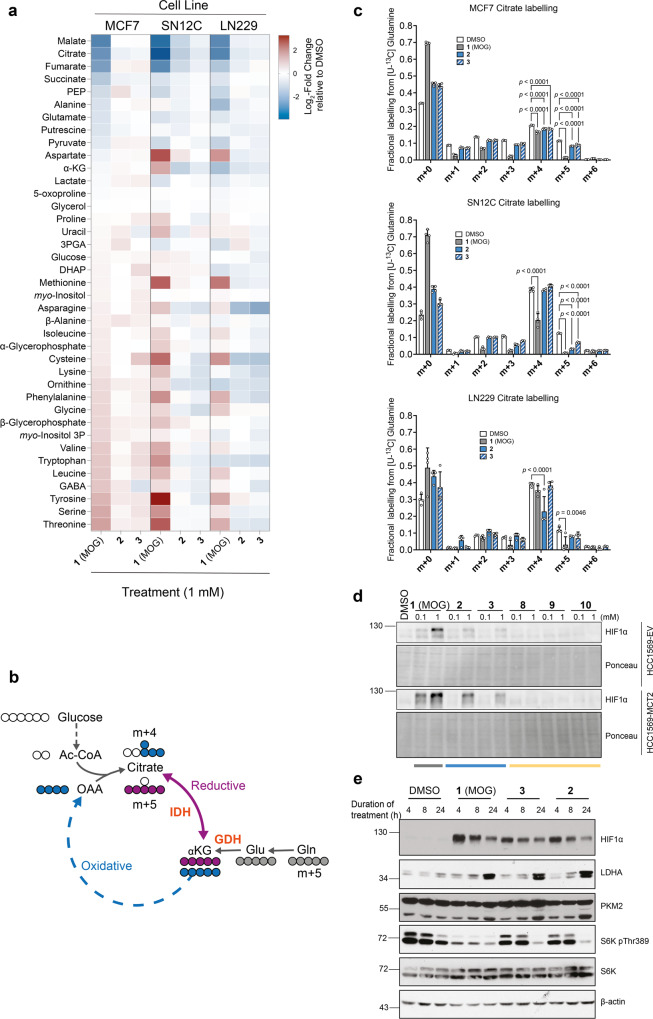
α-ketoglutarate (αKG) is a central metabolic node with a broad influence on cellular physiology. The αKG analogue N-oxalylglycine (NOG) and its membrane-permeable pro-drug derivative dimethyl-oxalylglycine (DMOG) have been extensively used as tools to study prolyl hydroxylases (PHDs) and other αKG-dependent processes. In cell culture media, DMOG is rapidly converted to MOG, which enters cells through monocarboxylate transporter MCT2, leading to intracellular NOG concentrations that are sufficiently high to inhibit glutaminolysis enzymes and cause cytotoxicity. Therefore, the degree of (D)MOG instability together with MCT2 expression levels determine the intracellular targets NOG engages with and, ultimately, its effects on cell viability. Here we designed and characterised a series of MOG analogues with the aims of improving compound stability and exploring the functional requirements for interaction with MCT2, a relatively understudied member of the SLC16 family. We report MOG analogues that maintain ability to enter cells via MCT2, and identify compounds that do not inhibit glutaminolysis or cause cytotoxicity but can still inhibit PHDs. We use these analogues to show that, under our experimental conditions, glutaminolysis-induced activation of mTORC1 can be uncoupled from PHD activity. Therefore, these new compounds can help deconvolute cellular effects that result from the polypharmacological action of NOG.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Woolley DW. The revolution in pharmacology. Perspect. Biol. Med. 1958;1:174–197. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
