

Reversal of the renal hyperglycemic memory in diabetic kidney disease by targeting sustained tubular p21 expression
- PMID: 36030260
- PMCID: PMC9420151
- DOI: 10.1038/s41467-022-32477-9
Reversal of the renal hyperglycemic memory in diabetic kidney disease by targeting sustained tubular p21 expression
Abstract
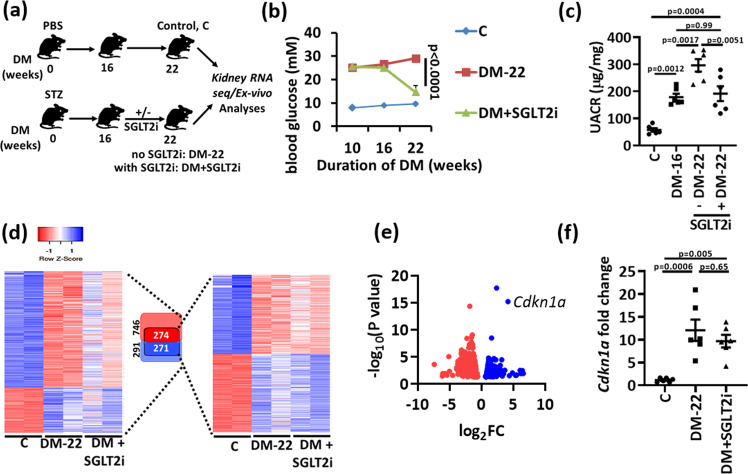
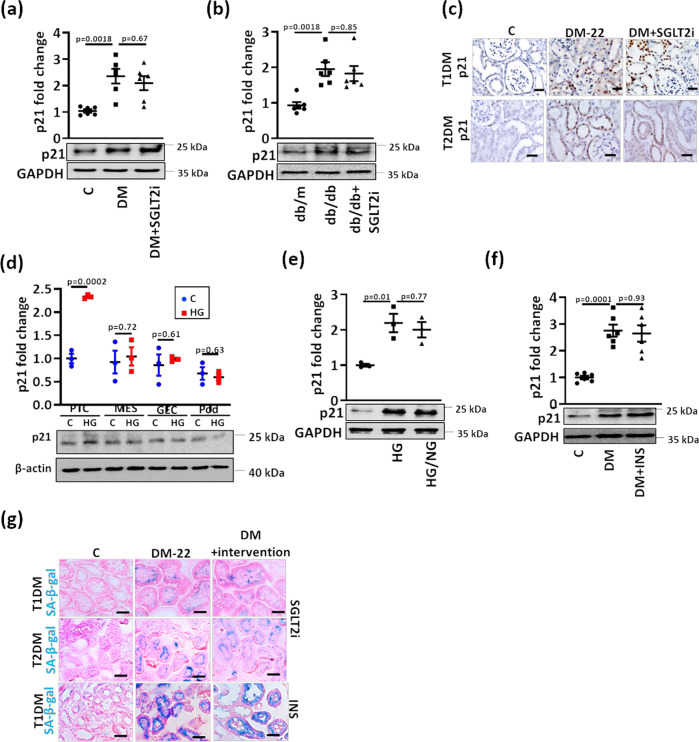
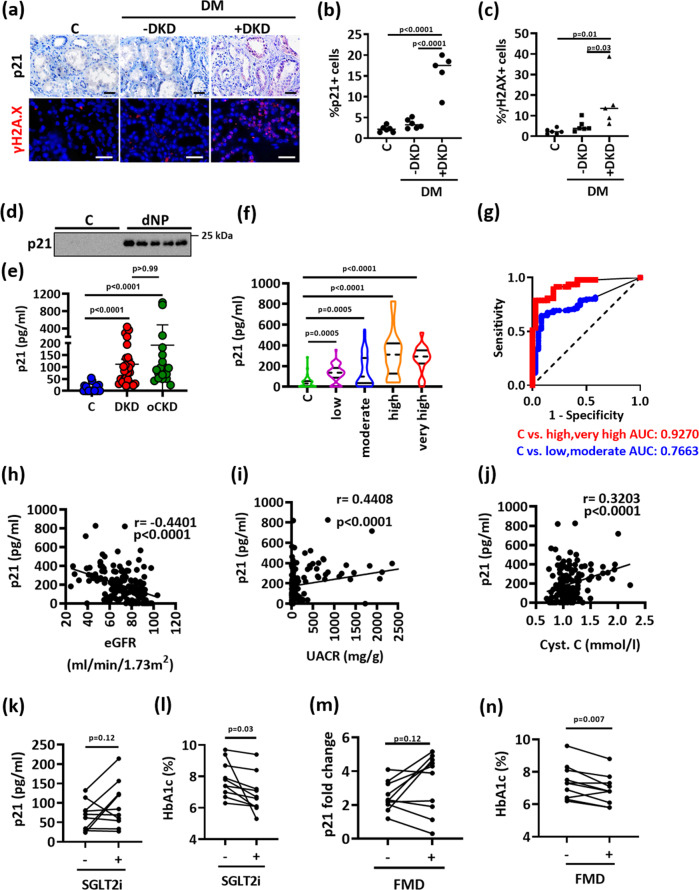
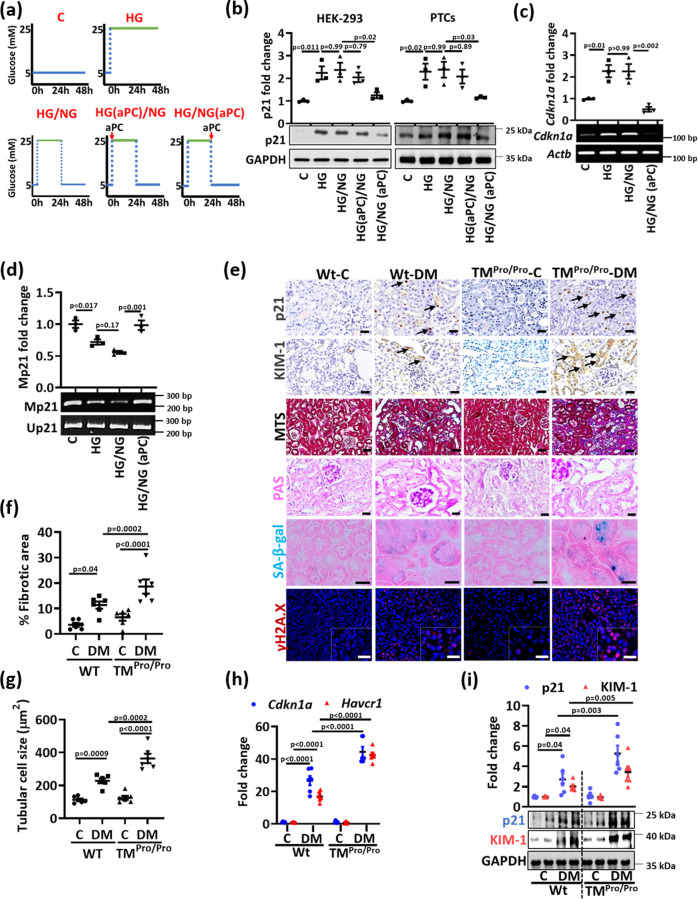
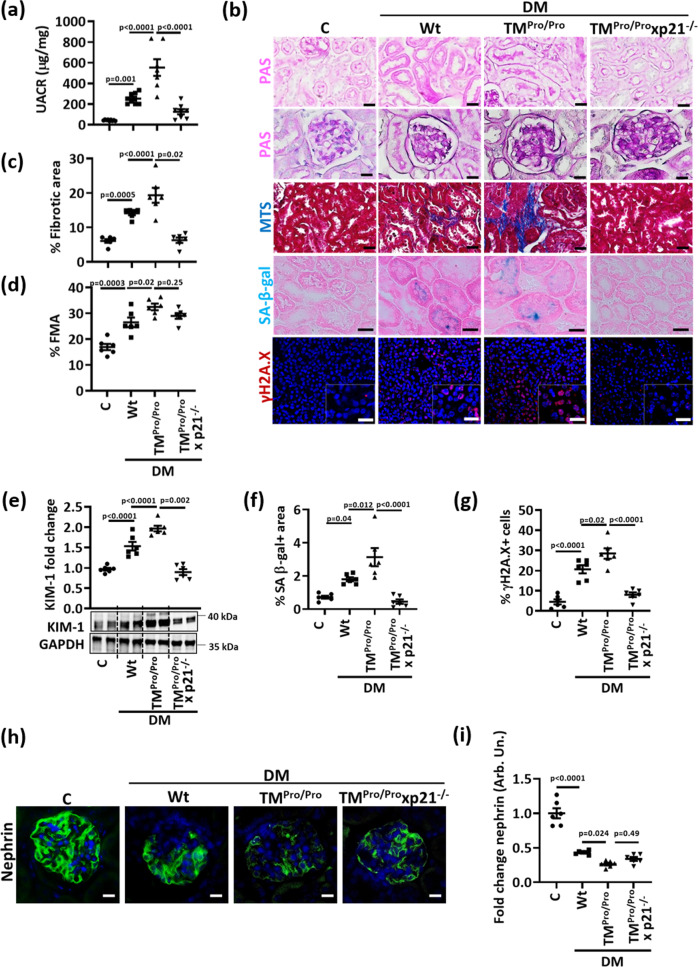
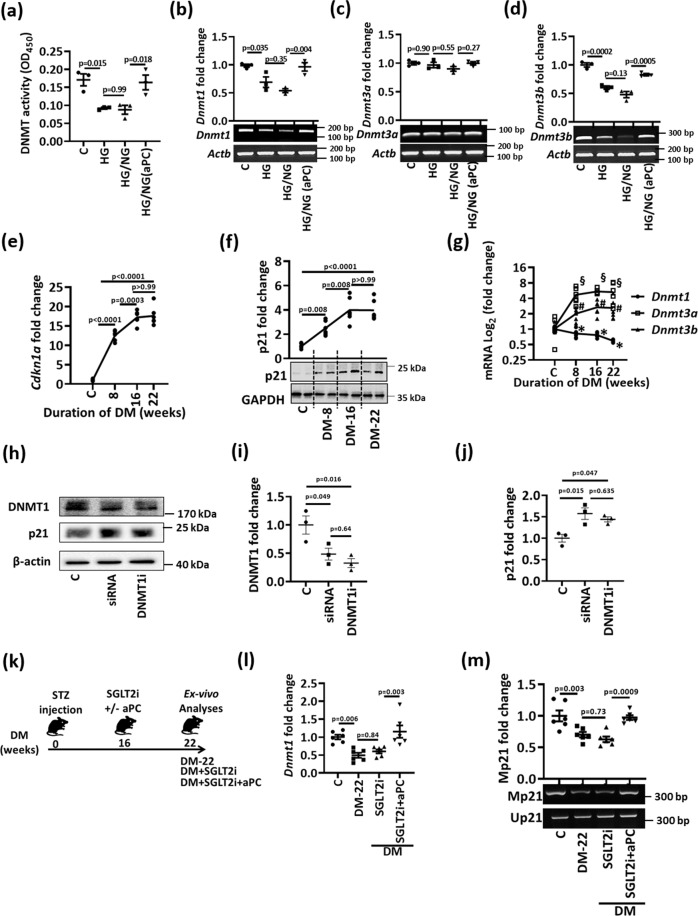
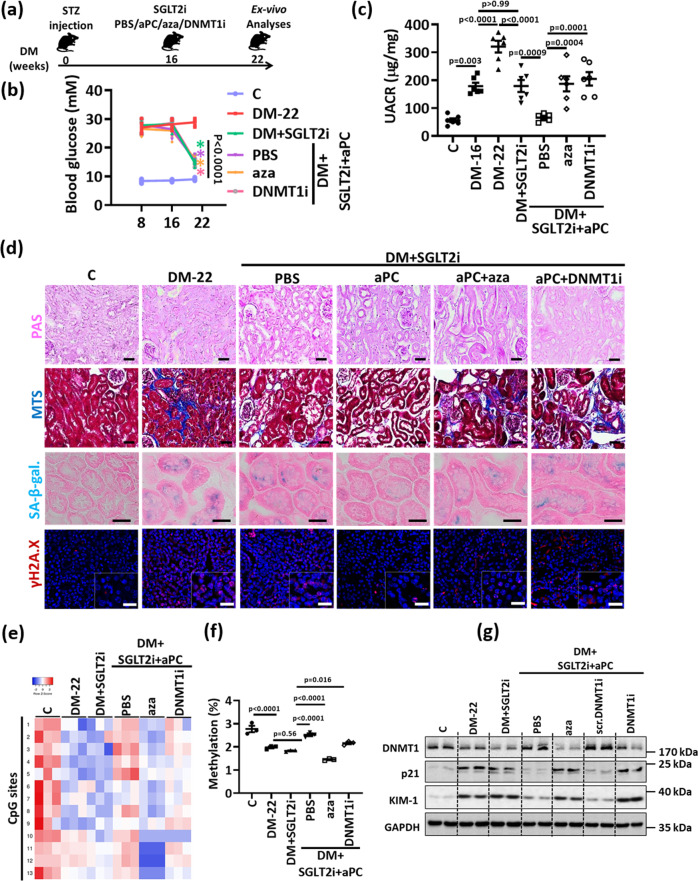
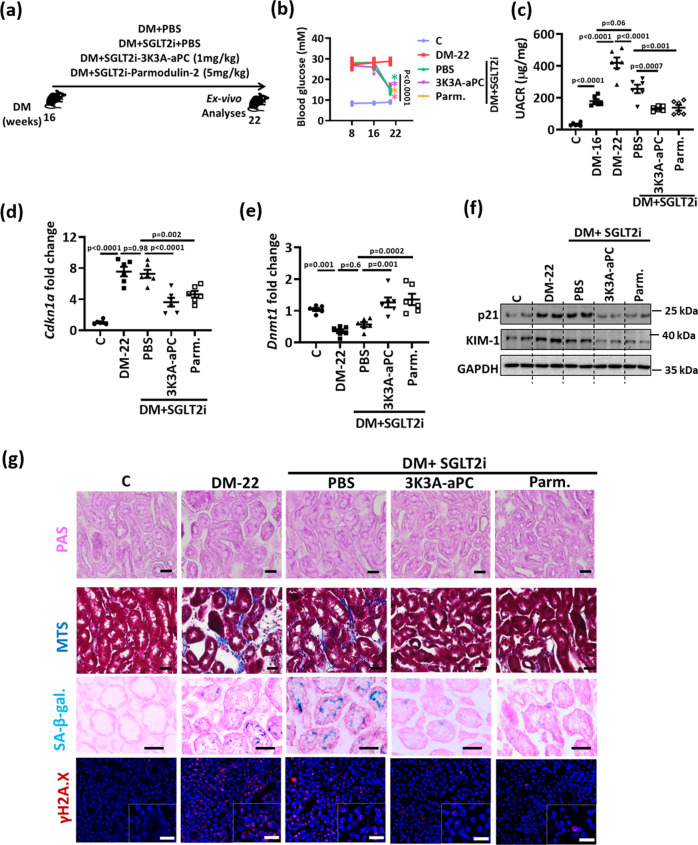
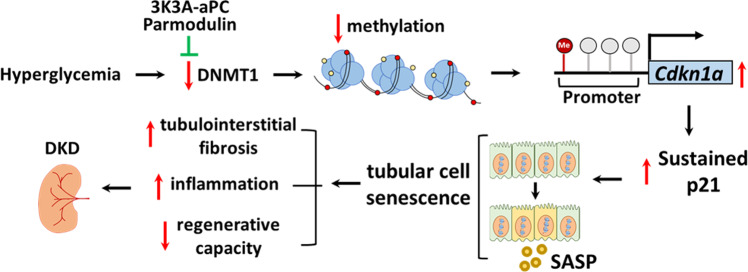
A major obstacle in diabetes is the metabolic or hyperglycemic memory, which lacks specific therapies. Here we show that glucose-mediated changes in gene expression largely persist in diabetic kidney disease (DKD) despite reversing hyperglycemia. The senescence-associated cyclin-dependent kinase inhibitor p21 (Cdkn1a) was the top hit among genes persistently induced by hyperglycemia and was associated with induction of the p53-p21 pathway. Persistent p21 induction was confirmed in various animal models, human samples and in vitro models. Tubular and urinary p21-levels were associated with DKD severity and remained elevated despite improved blood glucose levels in humans. Mechanistically, sustained tubular p21 expression in DKD is linked to demethylation of its promoter and reduced DNMT1 expression. Two disease resolving agents, protease activated protein C (3K3A-aPC) and parmodulin-2, reversed sustained tubular p21 expression, tubular senescence, and DKD. Thus, p21-dependent tubular senescence is a pathway contributing to the hyperglycemic memory, which can be therapeutically targeted.
© 2022. The Author(s).
Conflict of interest statement
C.D. is an inventor on patent applications and patents describing parmodulin compounds. The other authors declare no competing interests.
Figures
References
-
- Fouli, G. E. & Gnudi, L. The future: experimental therapies for renal disease in Diabetes. Nephron, 1–5, 10.1159/000492825 (2018). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
