

Novel role of macrophage TXNIP-mediated CYLD-NRF2-OASL1 axis in stress-induced liver inflammation and cell death
- PMID: 36035360
- PMCID: PMC9404660
- DOI: 10.1016/j.jhepr.2022.100532
Novel role of macrophage TXNIP-mediated CYLD-NRF2-OASL1 axis in stress-induced liver inflammation and cell death
Abstract
Background & aims: The stimulator of interferon genes (STING)/TANK-binding kinase 1 (TBK1) pathway is vital in mediating innate immune and inflammatory responses during oxidative/endoplasmic reticulum (ER) stress. However, it remains unknown whether macrophage thioredoxin-interacting protein (TXNIP) may regulate TBK1 function and cell death pathways during oxidative/ER stress.
Methods: A mouse model of hepatic ischaemia/reperfusion injury (IRI), the primary hepatocytes, and bone marrow-derived macrophages were used in the myeloid-specific TXNIP knockout (TXNIPM-KO) and TXNIP-proficient (TXNIPFL/FL) mice.
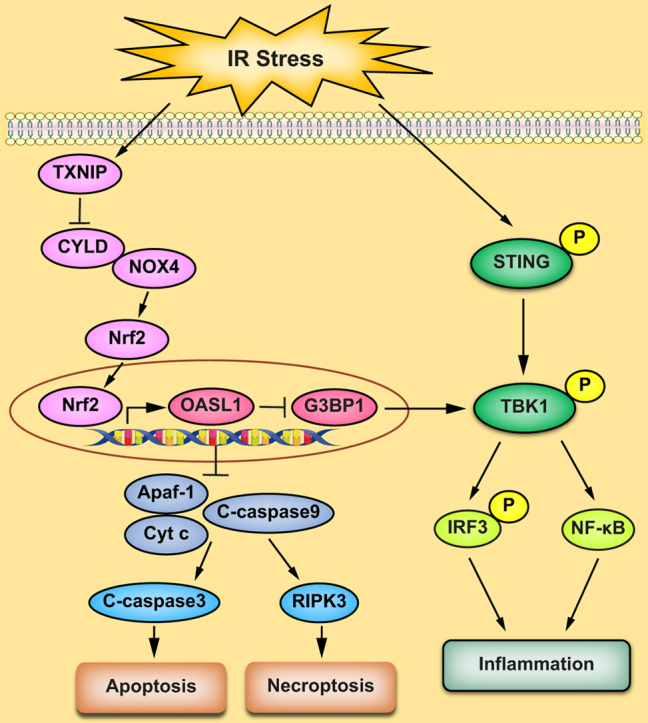
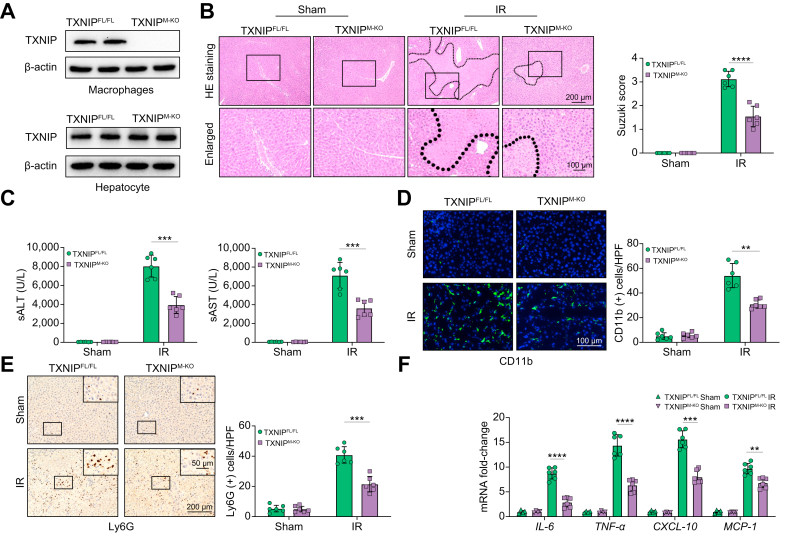
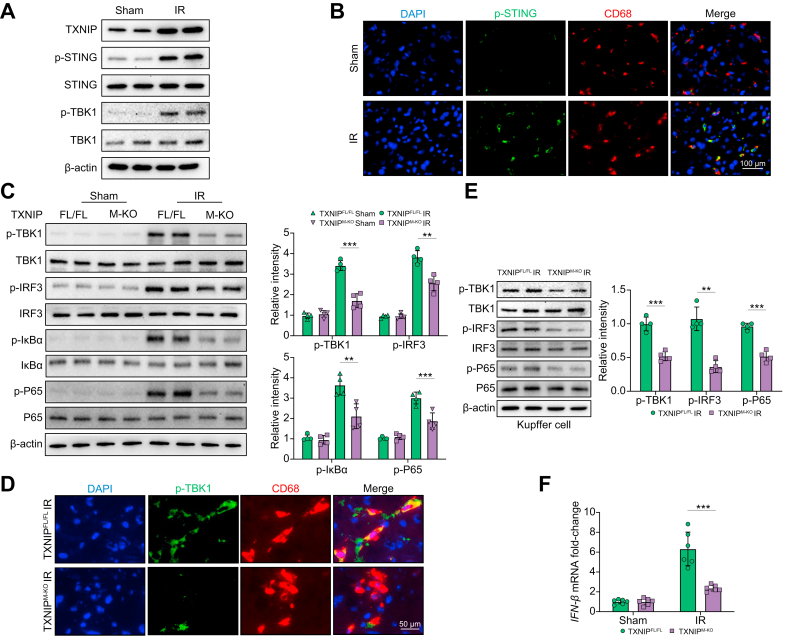
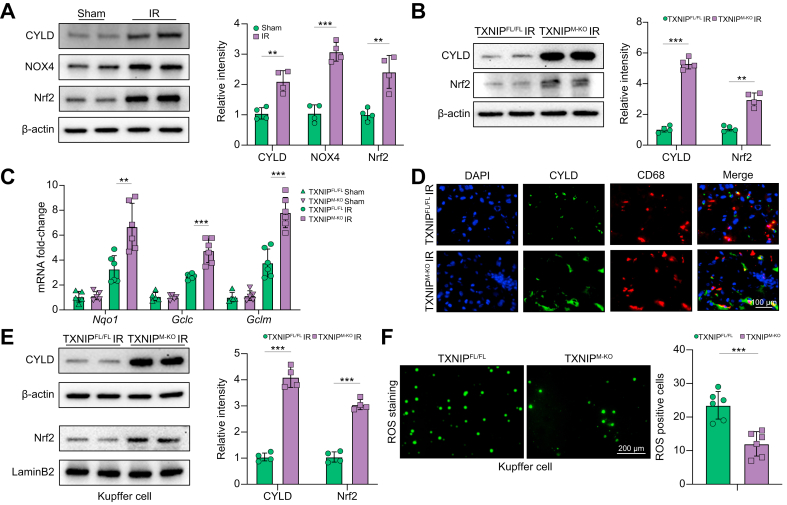
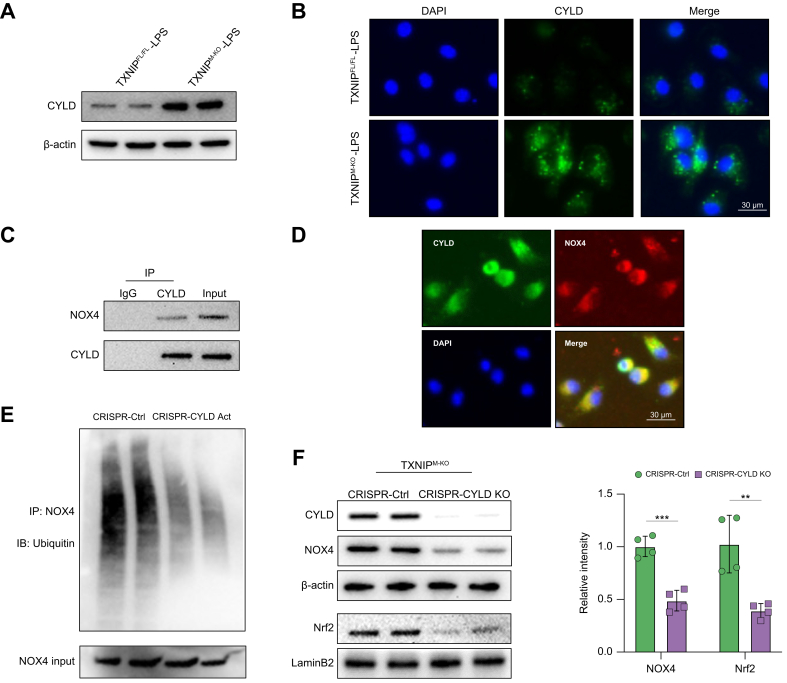
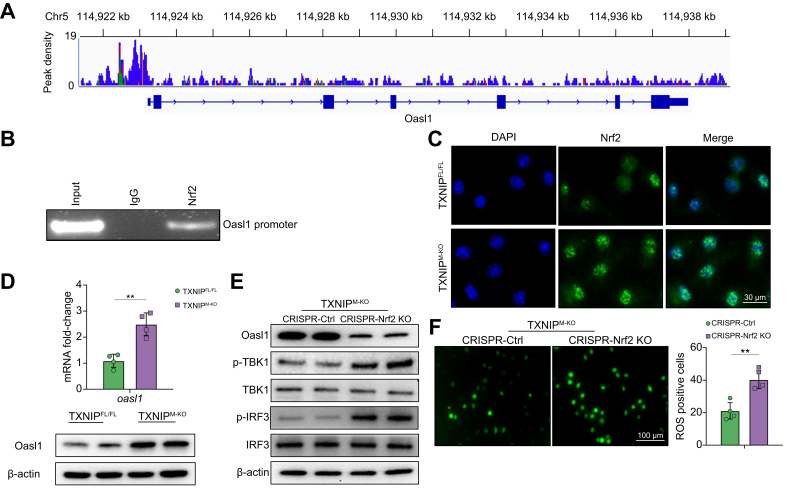
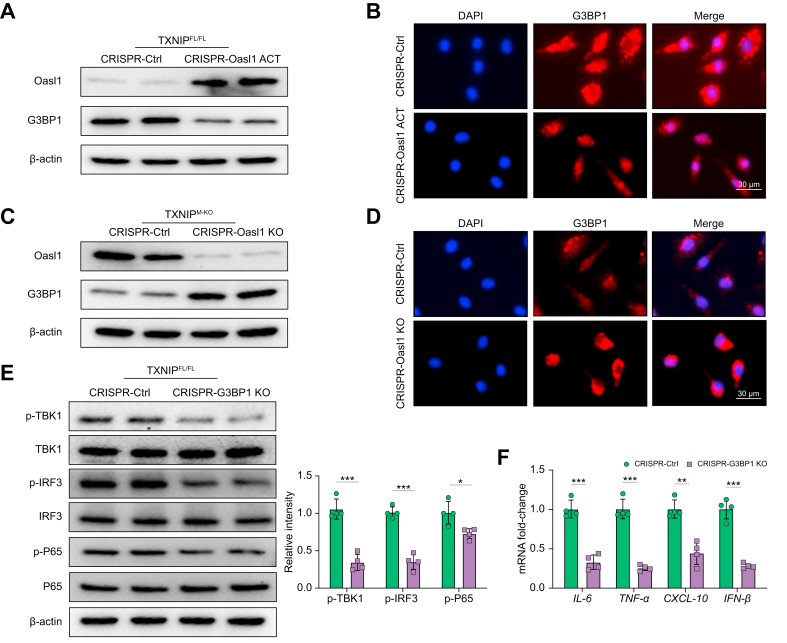
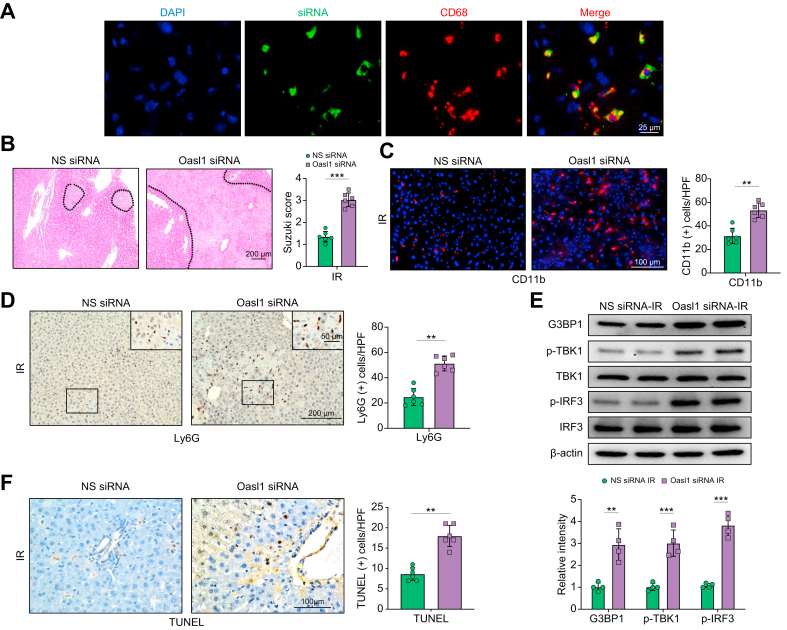
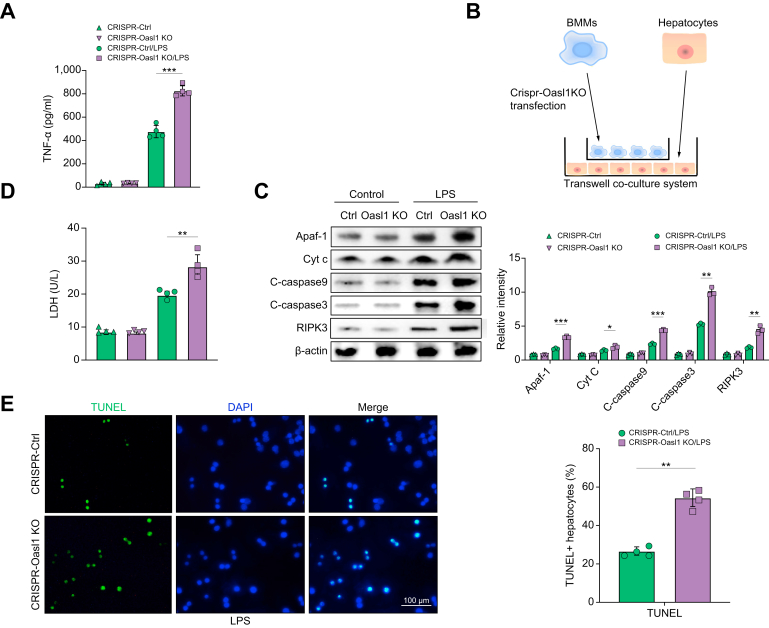
Results: The TXNIPM-KO mice were resistant to ischaemia/reperfusion (IR) stress-induced liver damage with reduced serum alanine aminotransferase (ALT)/aspartate aminotransferase (AST) levels, macrophage/neutrophil infiltration, and pro-inflammatory mediators compared with the TXNIPFL/FL controls. IR stress increased TXNIP, p-STING, and p-TBK1 expression in ischaemic livers. However, TXNIPM-KO inhibited STING, TBK1, interferon regulatory factor 3 (IRF3), and NF-κB activation with interferon-β (IFN-β) expression. Interestingly, TXNIPM-KO augmented nuclear factor (erythroid-derived 2)-like 2 (NRF2) activity, increased antioxidant gene expression, and reduced macrophage reactive oxygen species (ROS) production and hepatic apoptosis/necroptosis in IR-stressed livers. Mechanistically, macrophage TXNIP deficiency promoted cylindromatosis (CYLD), which colocalised and interacted with NADPH oxidase 4 (NOX4) to enhance NRF2 activity by deubiquitinating NOX4. Disruption of macrophage NRF2 or its target gene 2',5' oligoadenylate synthetase-like 1 (OASL1) enhanced Ras GTPase-activating protein-binding protein 1 (G3BP1) and TBK1-mediated inflammatory response. Notably, macrophage OASL1 deficiency induced hepatocyte apoptotic peptidase activating factor 1 (APAF1), cytochrome c, and caspase-9 activation, leading to increased caspase-3-initiated apoptosis and receptor-interacting serine/threonine-protein kinase 3 (RIPK3)-mediated necroptosis.
Conclusions: Macrophage TXNIP deficiency enhances CYLD activity and activates the NRF2-OASL1 signalling, controlling IR stress-induced liver injury. The target gene OASL1 regulated by NRF2 is crucial for modulating STING-mediated TBK1 activation and Apaf1/cytochrome c/caspase-9-triggered apoptotic/necroptotic cell death pathway. Our findings underscore a novel role of macrophage TXNIP-mediated CYLD-NRF2-OASL1 axis in stress-induced liver inflammation and cell death, implying the potential therapeutic targets in liver inflammatory diseases.
Lay summary: Liver inflammation and injury induced by ischaemia and reperfusion (the absence of blood flow to the liver tissue followed by the resupply of blood) is a significant cause of hepatic dysfunction and failure following liver transplantation, resection, and haemorrhagic shock. Herein, we uncover an underlying mechanism that contributes to liver inflammation and cell death in this setting and could be a therapeutic target in stress-induced liver inflammatory injury.
Keywords: ALT, alanine aminotransferase; APAF1, apoptotic peptidase activating factor 1; ASK1, apoptosis signal-regulating kinase 1; AST, aspartate aminotransferase; Apoptosis; BMM, bone marrow-derived macrophage; CXCL-10, C-X-C motif chemokine ligand 10; CYLD, cyclindromatosis; ChIP, chromatin immunoprecipitation; DAMP, damage-associated molecular pattern; DUB, deubiquitinating enzyme; ER, endoplasmic reticulum; ES, embryonic stem; G3BP1; G3BP1, Ras GTPase-activating protein-binding protein 1; GCLC, glutamate-cysteine ligase catalytic subunit; GCLM, glutamate-cysteine ligase regulatory subunit; IHC, immunohistochemistry; INF-β, interferon-β; IR, ischaemia/reperfusion; IRF3; IRF3, interferon regulatory factor 3; IRF7, IFN-regulating transcription factor 7; IRI, ischaemia/reperfusion injury; Innate immunity; KO, knockout; LPS, lipopolysaccharide; Liver inflammation; Lyz2, Lysozyme 2; MCP-1, monocyte chemoattractant protein 1; NOX2, NADPH oxidase 2; NOX4, NADPH oxidase 4; NQO1, NAD(P)H quinone dehydrogenase 1; NRF2, nuclear factor (erythroid-derived 2)-like 2; NS, non-specific; Necroptosis; OASL1, 2′,5′oligoadenylate synthetase-like 1; PAMP, pathogen-derived molecular pattern; RIPK3, receptor-interacting serine/threonine-protein kinase 3; ROS, reactive oxygen species; STING; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1; TLR4, Toll-like receptor 4; TNF-α, tumour necrosis factor-alpha; TRX, thioredoxin; TSS, transcription start sites; TXNIP, thioredoxin-interacting protein; TXNIPFL/FL, floxed TXNIP; TXNIPM-KO, myeloid-specific TXNIP KO; UTR, untranslated region; sALT, serum ALT; sAST, serum AST; siRNA, small interfering RNA.
© 2022 The Authors.
Conflict of interest statement
The authors declare no conflict of interest. Please refer to the accompanying ICMJE disclosure forms for further details.
Figures
References
-
- Soehnlein O., Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. 2010;10:427–439. - PubMed
-
- Lotze M.T., Zeh H.J., Rubartelli A., Sparvero L.J., Amoscato A.A., Washburn N.R., et al. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
