

A toolkit for enhanced reproducibility of RNASeq analysis for synthetic biologists
- PMID: 36035514
- PMCID: PMC9408027
- DOI: 10.1093/synbio/ysac012
A toolkit for enhanced reproducibility of RNASeq analysis for synthetic biologists
Abstract
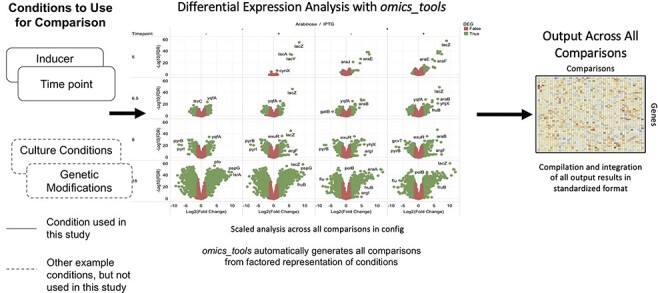
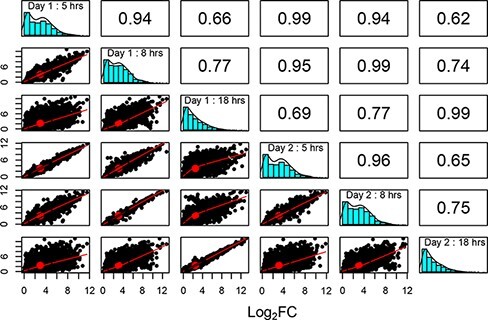
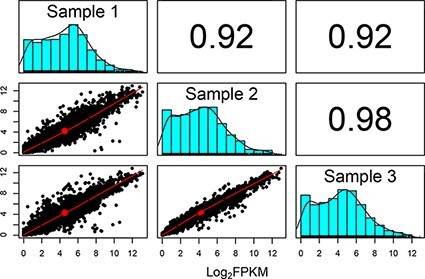
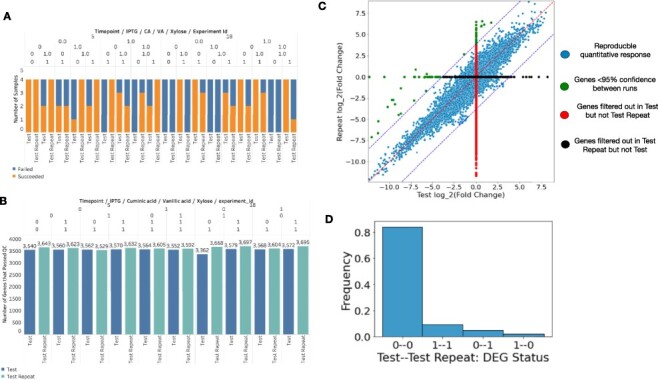
Sequencing technologies, in particular RNASeq, have become critical tools in the design, build, test and learn cycle of synthetic biology. They provide a better understanding of synthetic designs, and they help identify ways to improve and select designs. While these data are beneficial to design, their collection and analysis is a complex, multistep process that has implications on both discovery and reproducibility of experiments. Additionally, tool parameters, experimental metadata, normalization of data and standardization of file formats present challenges that are computationally intensive. This calls for high-throughput pipelines expressly designed to handle the combinatorial and longitudinal nature of synthetic biology. In this paper, we present a pipeline to maximize the analytical reproducibility of RNASeq for synthetic biologists. We also explore the impact of reproducibility on the validation of machine learning models. We present the design of a pipeline that combines traditional RNASeq data processing tools with structured metadata tracking to allow for the exploration of the combinatorial design in a high-throughput and reproducible manner. We then demonstrate utility via two different experiments: a control comparison experiment and a machine learning model experiment. The first experiment compares datasets collected from identical biological controls across multiple days for two different organisms. It shows that a reproducible experimental protocol for one organism does not guarantee reproducibility in another. The second experiment quantifies the differences in experimental runs from multiple perspectives. It shows that the lack of reproducibility from these different perspectives can place an upper bound on the validation of machine learning models trained on RNASeq data. Graphical Abstract.
Keywords: automation; machine learning; standardization; transcriptomics.
© The Author(s) 2022. Published by Oxford University Press.
Figures

References
-
- Babraham Bioinformatics – FastQC: A quality control tool for high throughput sequence data . https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (8 August 2004, date last accessed).
-
- Picard Toolkit . (2019) Broad Institute, GitHub Repository. https://broadinstitute.github.io/picard/.
-
- Brookes E. and Stubbs J. (2019) GenApp, containers and Abaco: technical paper. In: Proceedings of the Practice and Experience in Advanced Research Computing on Rise of the Machines (Learning). Association for Computing Machinery, New York, NY, USA, pp. 1–8.
LinkOut - more resources
Full Text Sources