

The role of C-terminal helix in the conformational transition of an arginine binding protein
- PMID: 36035778
- PMCID: PMC9402392
- DOI: 10.1016/j.yjsbx.2022.100071
The role of C-terminal helix in the conformational transition of an arginine binding protein
Abstract
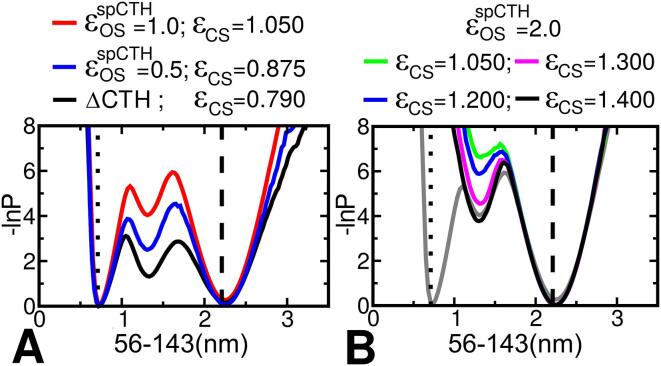
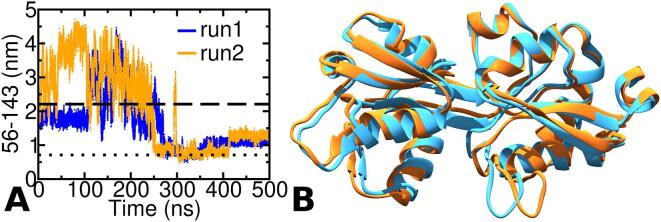
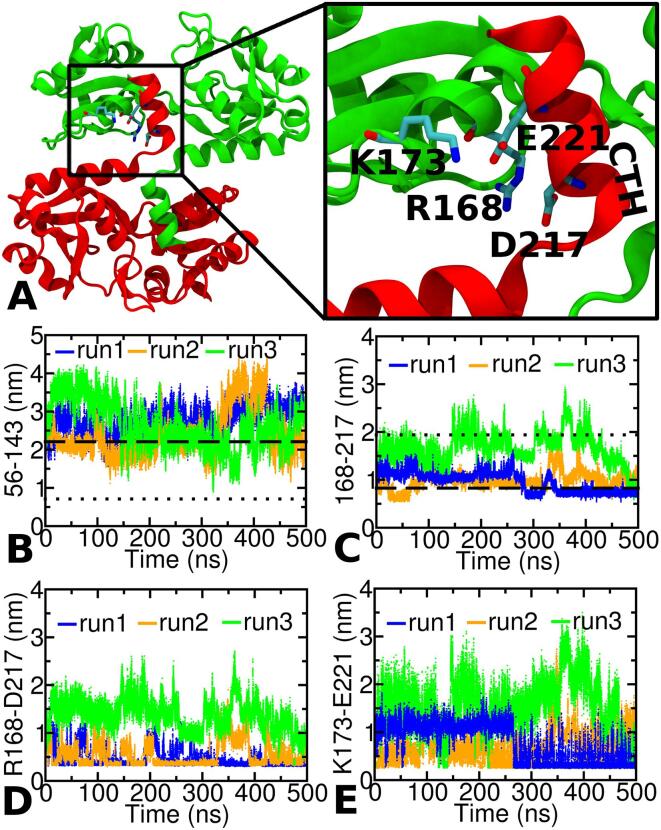
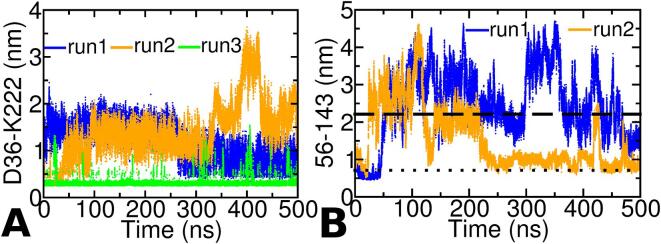
The thermotoga maritima arginine binding protein (TmArgBP) is a periplasmic binding protein that has a short helix at the C-terminal end (CTH), which is swapped between the two chains. We apply a coarse-grained structure-based model (SBM) and all-atom MD simulation on this protein to understand the mechanism and the role of CTH in the conformational transition. When the results of SBM simulations of TmArgBP in the presence and absence of CTH are compared, we find that CTH is strategically located at the back of the binding pocket restraining the open-state conformation thereby disengaging access to the closed-state. We also ran all-atom MD simulations of open-state TmArgBP with and without CTH and discovered that in the absence of CTH the protein could reach the closed-state within 250 ns, while in its presence, the protein remained predominantly in its open-state conformation. In the simulation started from unliganded closed-state conformation without CTH, the protein exhibited multiple transitions between the two states, suggesting CTH as an essential structural element to stabilize the open-state conformation. In another simulation that began with an unliganded closed-state conformation with CTH, the protein was able to access the open-state. In this simulation the CTH was observed to reorient itself to interact with the protein emphasizing its role in assisting the conformational change. Based on our findings, we believe that CTH not only acts as a structural element that constraints the protein in its open-state but it may also guide the protein back to its open-state conformation upon ligand unbinding.
Keywords: Arginine binding protein; C-terminal helix; CTH, C-terminal helix; Conformational transition; MD simulation; SBM, structure-based model; Structure-based model; TmArgBP, thermotoga maritima arginine binding protein.
© 2022 The Author(s).
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures


References
-
- Abraham M.J., Murtola T., Schulz R., Páll S., Smith J.C., Hess B., Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19–25. doi: 10.1016/j.softx.2015.06.001. - DOI
-
- Ausili A., Pennacchio A., Staiano M., Dattelbaum J.D., Fessas D., Schiraldi A., D’Auria S. Amino acid transport in thermophiles: characterization of an arginine-binding protein from Thermotoga maritima. 3. Conformational dynamics and stability. J. Photochem. Photobiol. B. 2013;118:66–73. doi: 10.1016/j.jphotobiol.2012.11.004. - DOI - PubMed
-
- Balasco N., Smaldone G., Vigorita M., Del Vecchio P., Graziano G., Ruggiero A., Vitagliano L. The characterization of thermotoga maritima arginine binding protein variants demonstrates that minimal local strains have an important impact on protein stability. Sci. Rep. 2019;9:6617. doi: 10.1038/s41598-019-43157-y. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
