

Multiscale fluorescence imaging of living samples
- PMID: 36036808
- PMCID: PMC9512758
- DOI: 10.1007/s00418-022-02147-4
Multiscale fluorescence imaging of living samples
Abstract
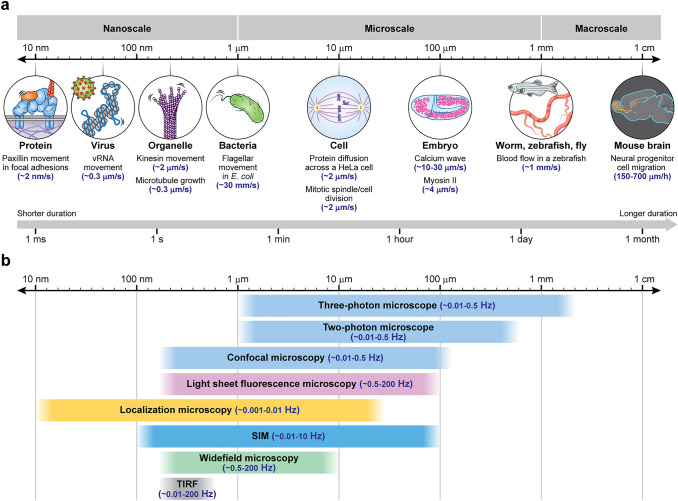
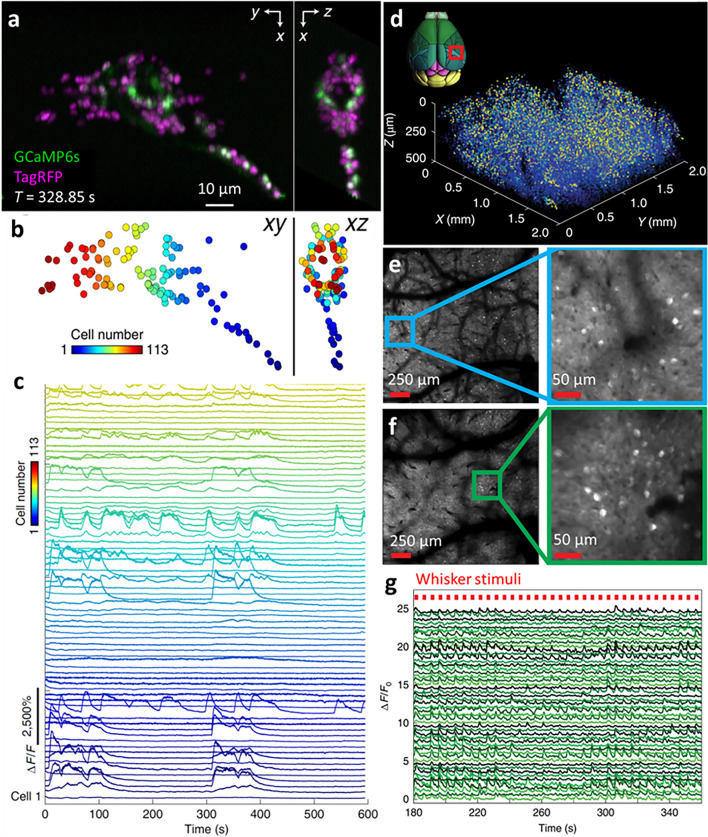
Fluorescence microscopy is a highly effective tool for interrogating biological structure and function, particularly when imaging across multiple spatiotemporal scales. Here we survey recent innovations and applications in the relatively understudied area of multiscale fluorescence imaging of living samples. We discuss fundamental challenges in live multiscale imaging and describe successful examples that highlight the power of this approach. We attempt to synthesize general strategies from these test cases, aiming to help accelerate progress in this exciting area.
Keywords: Fluorescence; Live imaging; Microscopy; Multiscale.
© 2022. This is a U.S. Government work and not under copyright protection in the US; foreign copyright protection may apply.
Conflict of interest statement
The authors declare no competing interests.
The NIH and its staff do not recommend or endorse any company, product, or service.
Figures







References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
