

Mitochondrial outer membrane protein FUNDC2 promotes ferroptosis and contributes to doxorubicin-induced cardiomyopathy
- PMID: 36037337
- PMCID: PMC9457330
- DOI: 10.1073/pnas.2117396119
Mitochondrial outer membrane protein FUNDC2 promotes ferroptosis and contributes to doxorubicin-induced cardiomyopathy
Abstract
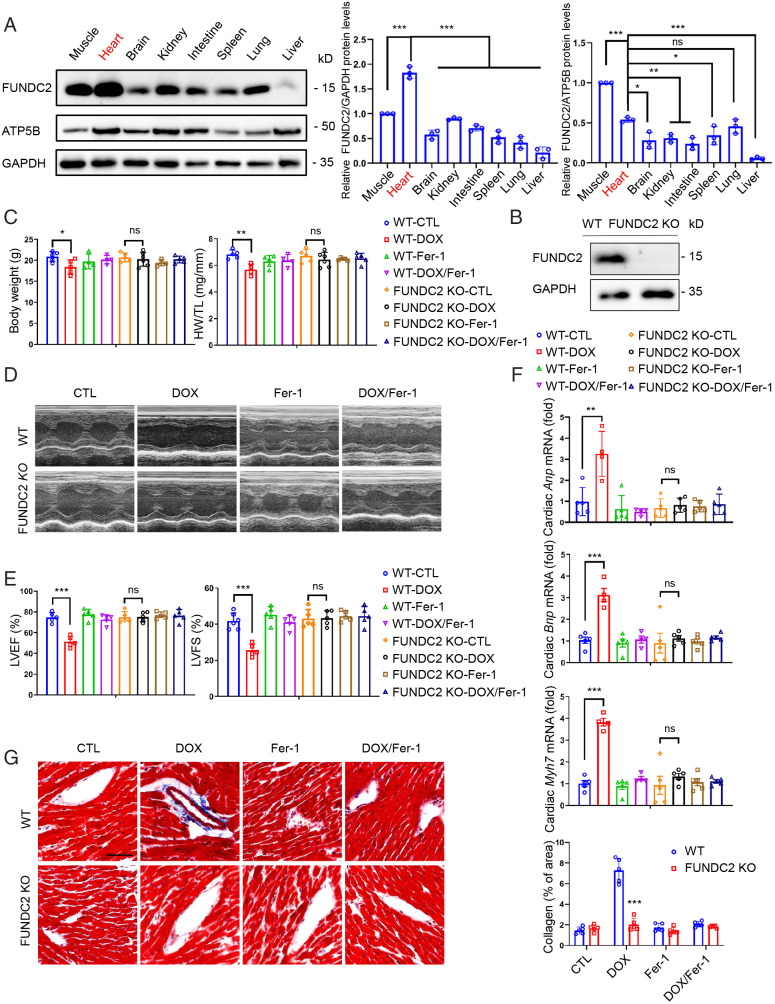
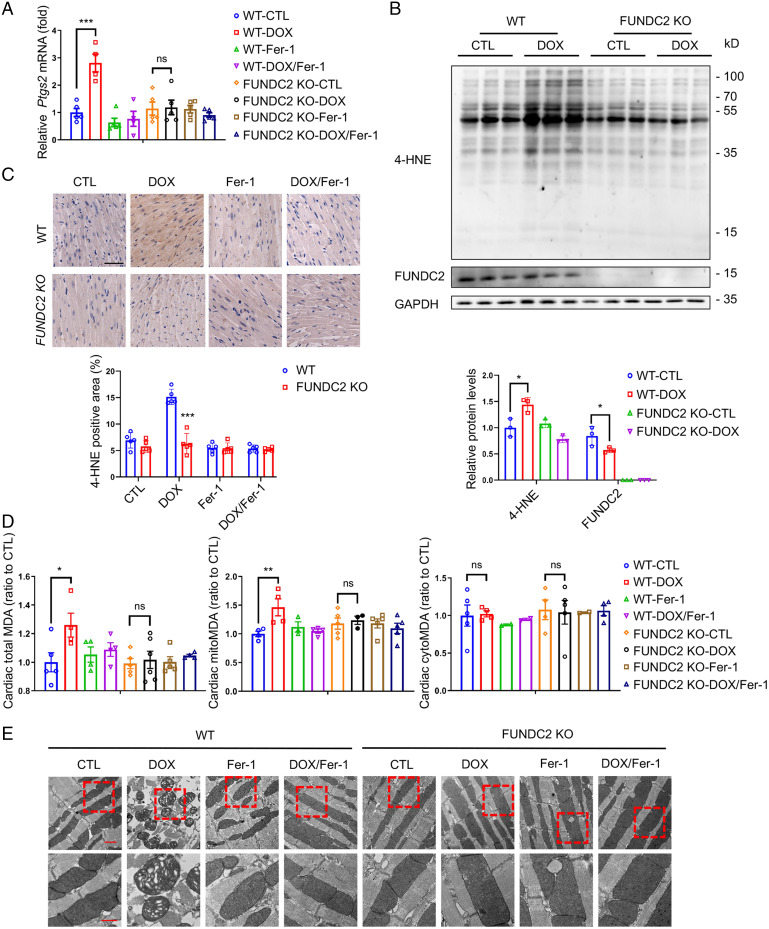
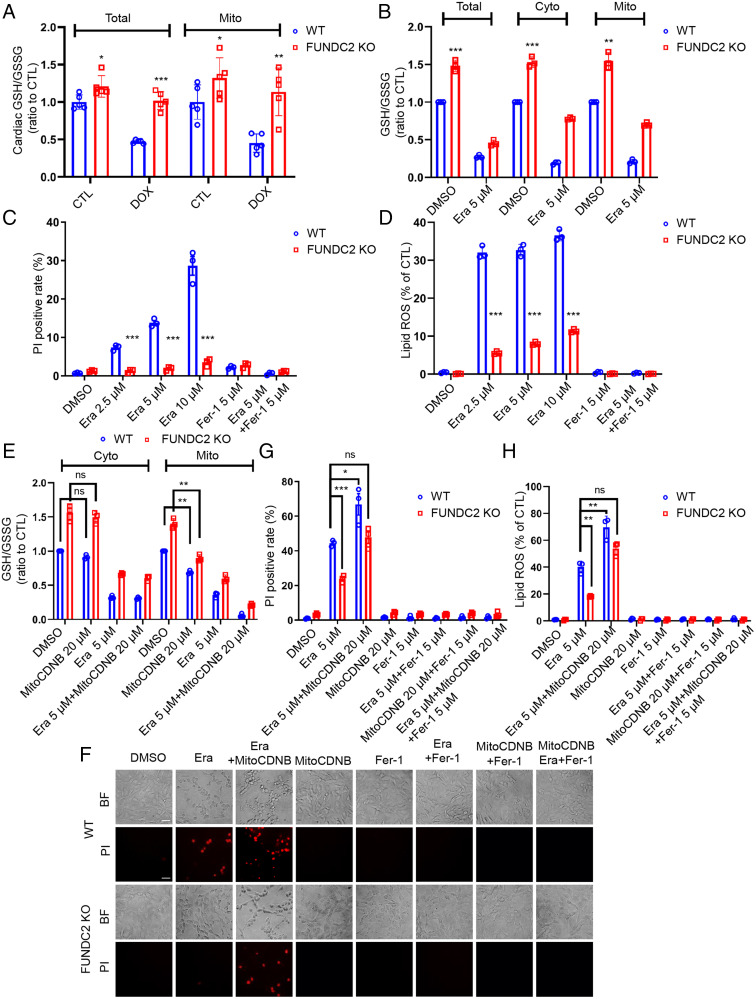
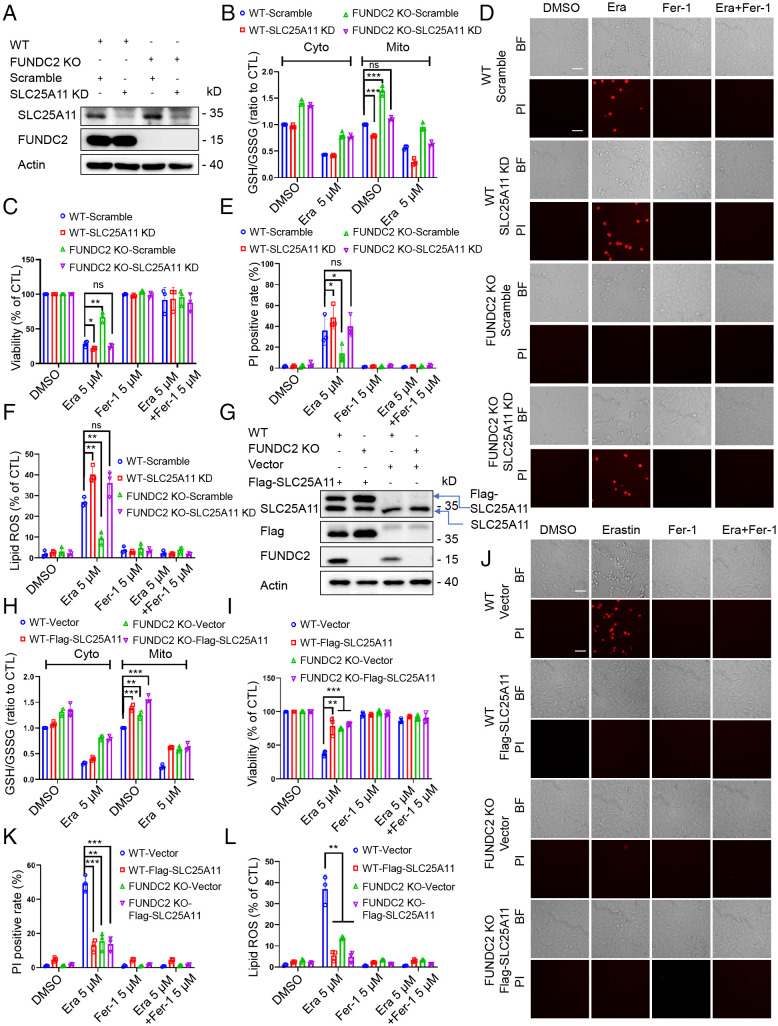
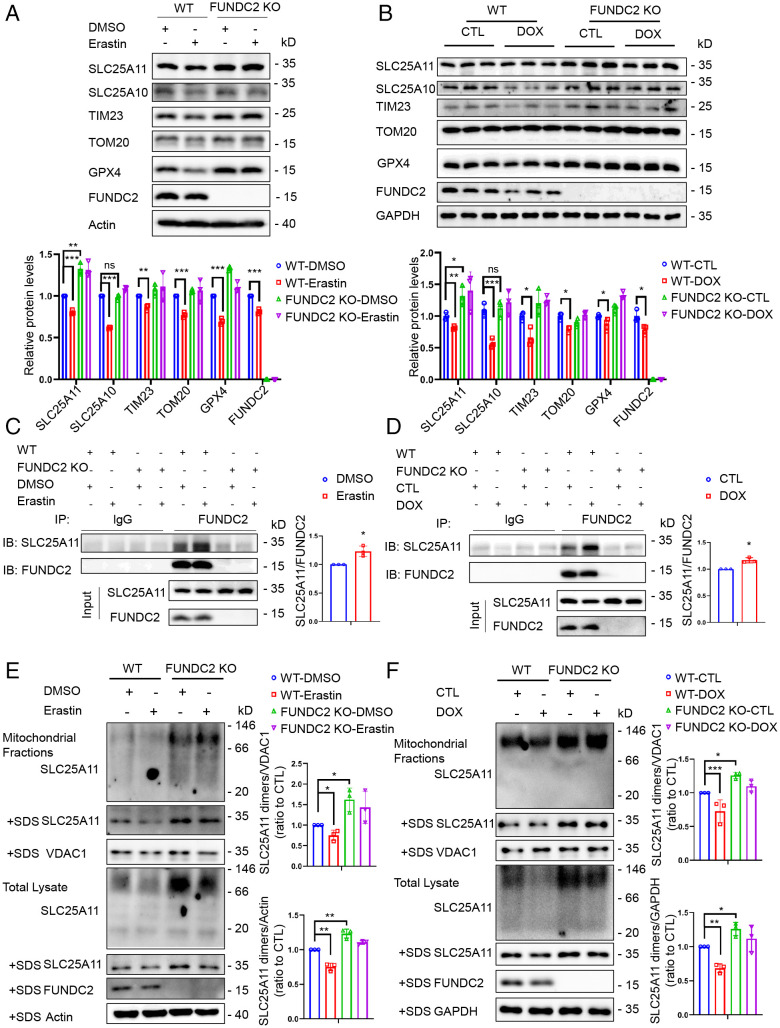
Ferroptosis is an iron-dependent programmed necrosis characterized by glutathione (GSH) depletion and lipid peroxidation (LPO). Armed with both the pro- and antiferroptosis machineries, mitochondria play a central role in ferroptosis. However, how mitochondria sense the stress to activate ferroptosis under (patho-)physiological settings remains incompletely understood. Here, we show that FUN14 domain-containing 2, also known as HCBP6 (FUNDC2), a highly conserved and ubiquitously expressed mitochondrial outer membrane protein, regulates ferroptosis and contributes to doxorubicin (DOX)-induced cardiomyopathy. We showed that knockout of FUNDC2 protected mice from DOX-induced cardiac injury by preventing ferroptosis. Mechanistic studies reveal that FUNDC2 interacts with SLC25A11, the mitochondrial glutathione transporter, to regulate mitoGSH levels. Specifically, knockdown of SLC25A11 in FUNDC2-knockout (KO) cells reduced mitoGSH and augmented erasin-induced ferroptosis. FUNDC2 also affected the stability of both SLC25A11 and glutathione peroxidase 4 (GPX4), key regulators for ferroptosis. Our results demonstrate that FUNDC2 modulates ferroptotic stress via regulating mitoGSH and further support a therapeutic strategy of cardioprotection by preventing mitoGSH depletion and ferroptosis.
Keywords: FUNDC2; SLC25A11; ferroptosis; mitoGSH; mitochondria.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
