

3D chromatin remodeling potentiates transcriptional programs driving cell invasion
- PMID: 36037342
- PMCID: PMC9457068
- DOI: 10.1073/pnas.2203452119
3D chromatin remodeling potentiates transcriptional programs driving cell invasion
Abstract
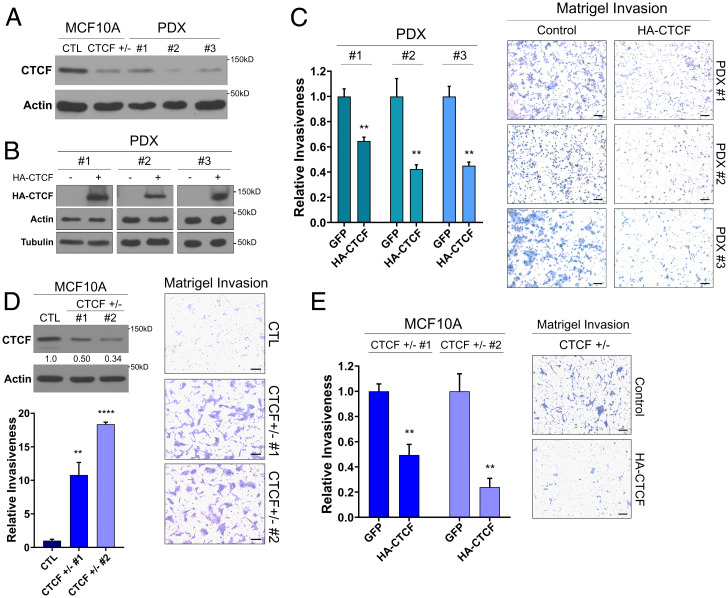
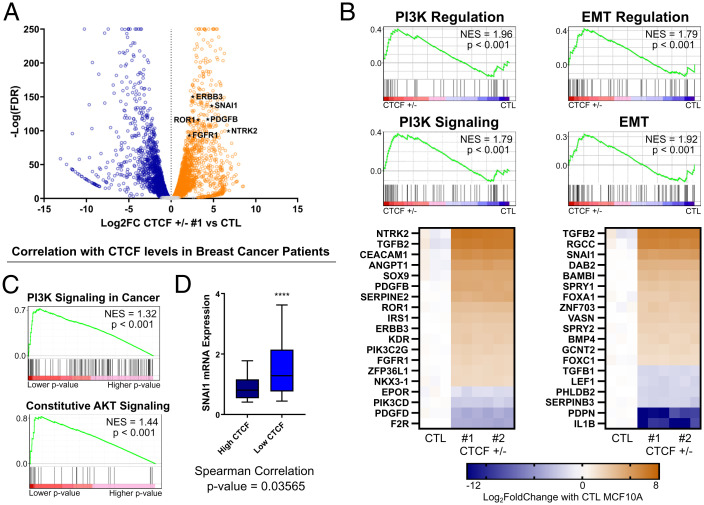
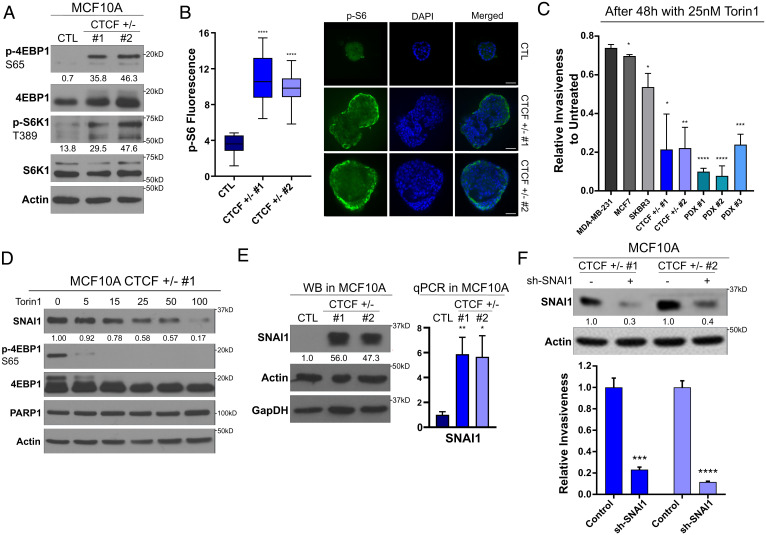
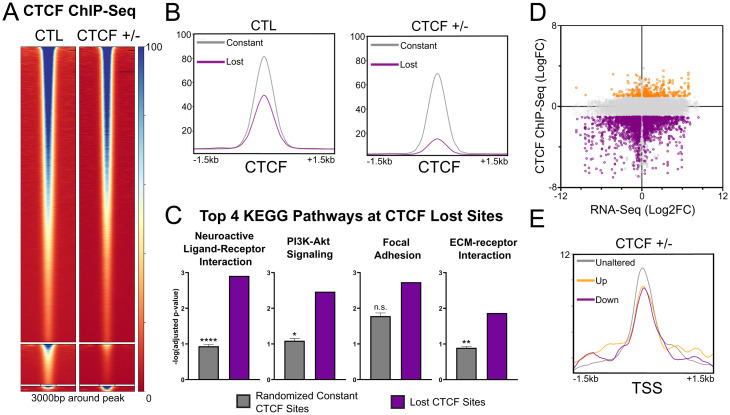
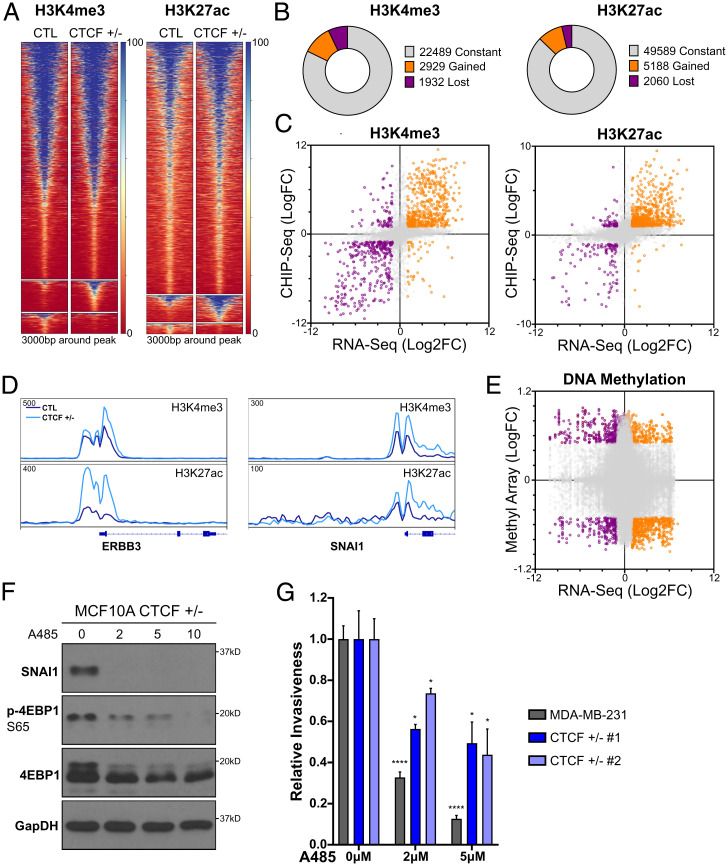
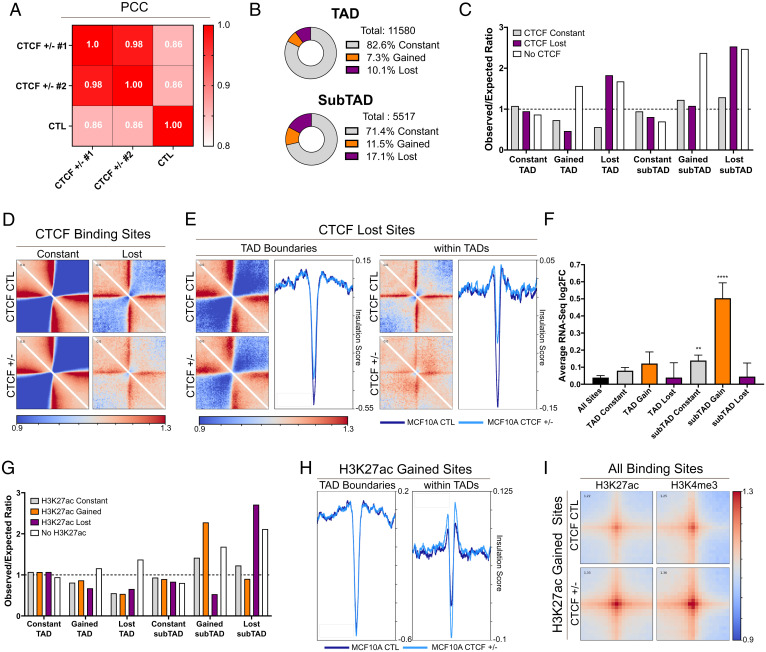
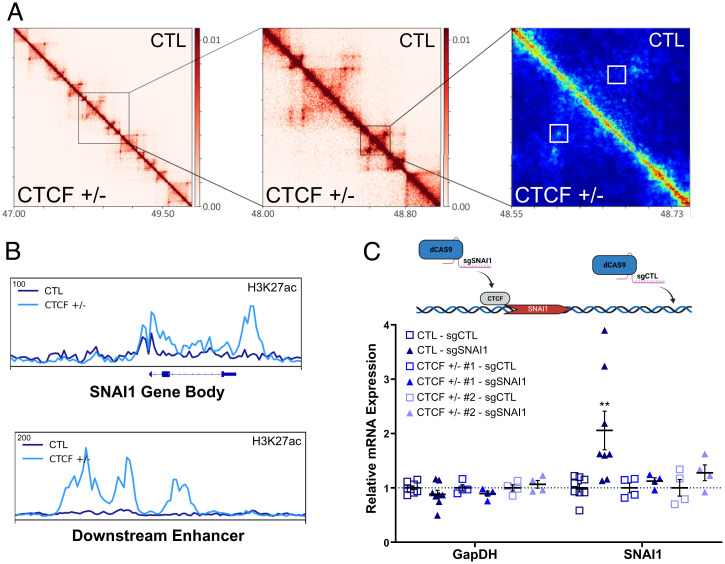
The contribution of deregulated chromatin architecture, including topologically associated domains (TADs), to cancer progression remains ambiguous. CCCTC-binding factor (CTCF) is a central regulator of higher-order chromatin structure that undergoes copy number loss in over half of all breast cancers, but the impact of this defect on epigenetic programming and chromatin architecture remains unclear. We find that under physiological conditions, CTCF organizes subTADs to limit the expression of oncogenic pathways, including phosphatidylinositol 3-kinase (PI3K) and cell adhesion networks. Loss of a single CTCF allele potentiates cell invasion through compromised chromatin insulation and a reorganization of chromatin architecture and histone programming that facilitates de novo promoter-enhancer contacts. However, this change in the higher-order chromatin landscape leads to a vulnerability to inhibitors of mTOR. These data support a model whereby subTAD reorganization drives both modification of histones at de novo enhancer-promoter contacts and transcriptional up-regulation of oncogenic transcriptional networks.
Keywords: CTCF; TAD; breast cancer; epigenetics; subTAD.
Conflict of interest statement
The authors declare no competing interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
