

Adaptive exchange sustains cullin-RING ubiquitin ligase networks and proper licensing of DNA replication
- PMID: 36037385
- PMCID: PMC9456757
- DOI: 10.1073/pnas.2205608119
Adaptive exchange sustains cullin-RING ubiquitin ligase networks and proper licensing of DNA replication
Abstract
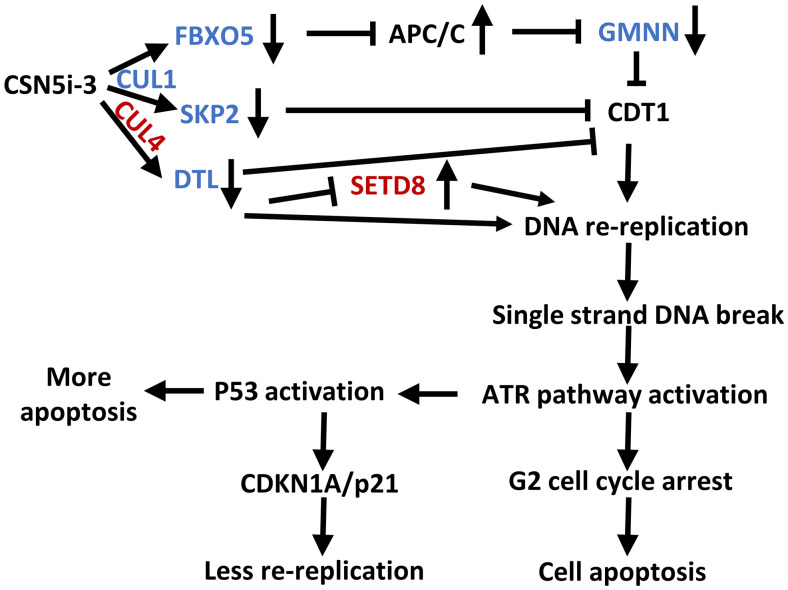
Cop9 signalosome (CSN) regulates the function of cullin-RING E3 ubiquitin ligases (CRLs) by deconjugating the ubiquitin-like protein NEDD8 from the cullin subunit. To understand the physiological impact of CSN function on the CRL network and cell proliferation, we combined quantitative mass spectrometry and genome-wide CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa) screens to identify factors that modulate cell viability upon inhibition of CSN by the small molecule CSN5i-3. CRL components and regulators strongly modulated the antiproliferative effects of CSN5i-3, and in addition we found two pathways involved in genome integrity, SCFFBXO5-APC/C-GMNN and CUL4DTL-SETD8, that contribute substantially to the toxicity of CSN inhibition. Our data highlight the importance of CSN-mediated NEDD8 deconjugation and adaptive exchange of CRL substrate receptors in sustaining CRL function and suggest approaches for leveraging CSN inhibition for the treatment of cancer.
Keywords: CRISPR screen; CSN5i-3; Cop9 signalosome; DNA replication; deneddylation.
Conflict of interest statement
Competing interest statement: Y.Z. is an employee of Amgen. D.L., C.-M. L., and R.J.D. are employees and shareholders of Amgen. J.L. was an employee of Amgen. J.R.L. was an employee and shareholder of Amgen. M.J. consults for Maze Therapeutics and Gate Biosciences. S.D.G. is the founder, president, CEO, and CTO of Proteas Bioanalytics, Inc. The work described in the paper does not have any direct financial implications for Amgen.
Figures






References
-
- Cardozo T., Pagano M., The SCF ubiquitin ligase: Insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 5, 739–751 (2004). - PubMed
-
- Willems A. R., Schwab M., Tyers M., A hitchhiker’s guide to the cullin ubiquitin ligases: SCF and its kin. Biochim. Biophys. Acta 1695, 133–170 (2004). - PubMed
-
- Deshaies R. J., Joazeiro C. A., RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 (2009). - PubMed
-
- Soucy T. A., et al. , An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736 (2009). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
