

Large-scale sequencing identifies multiple genes and rare variants associated with Crohn's disease susceptibility
- PMID: 36038634
- PMCID: PMC9700438
- DOI: 10.1038/s41588-022-01156-2
Large-scale sequencing identifies multiple genes and rare variants associated with Crohn's disease susceptibility
Abstract
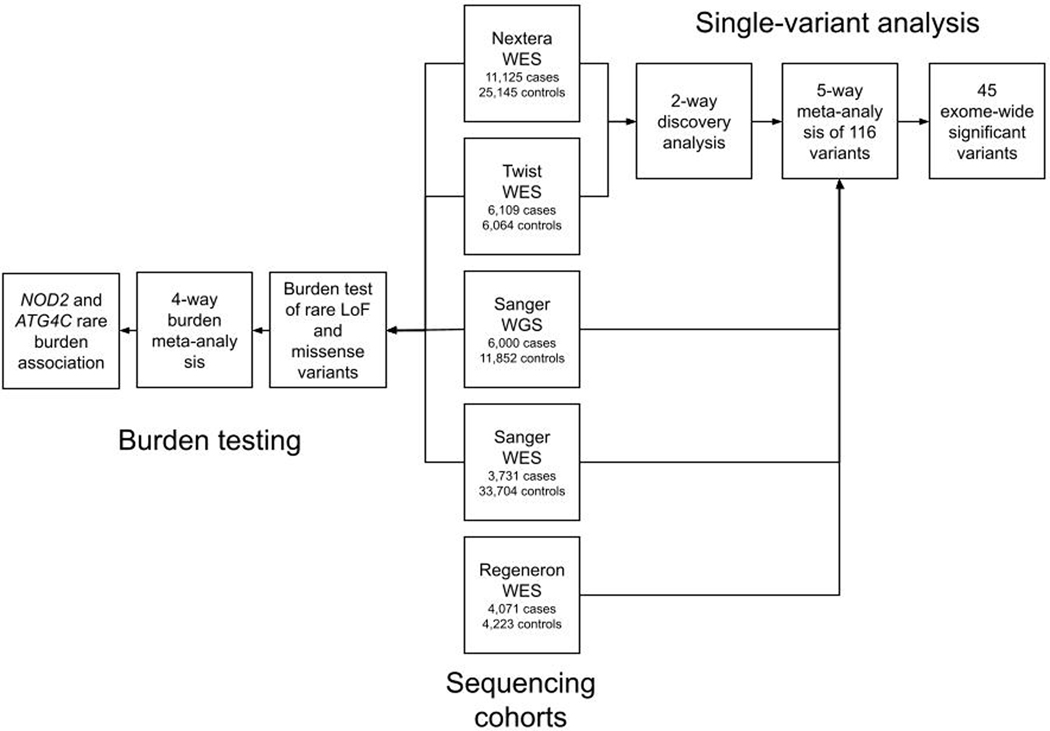
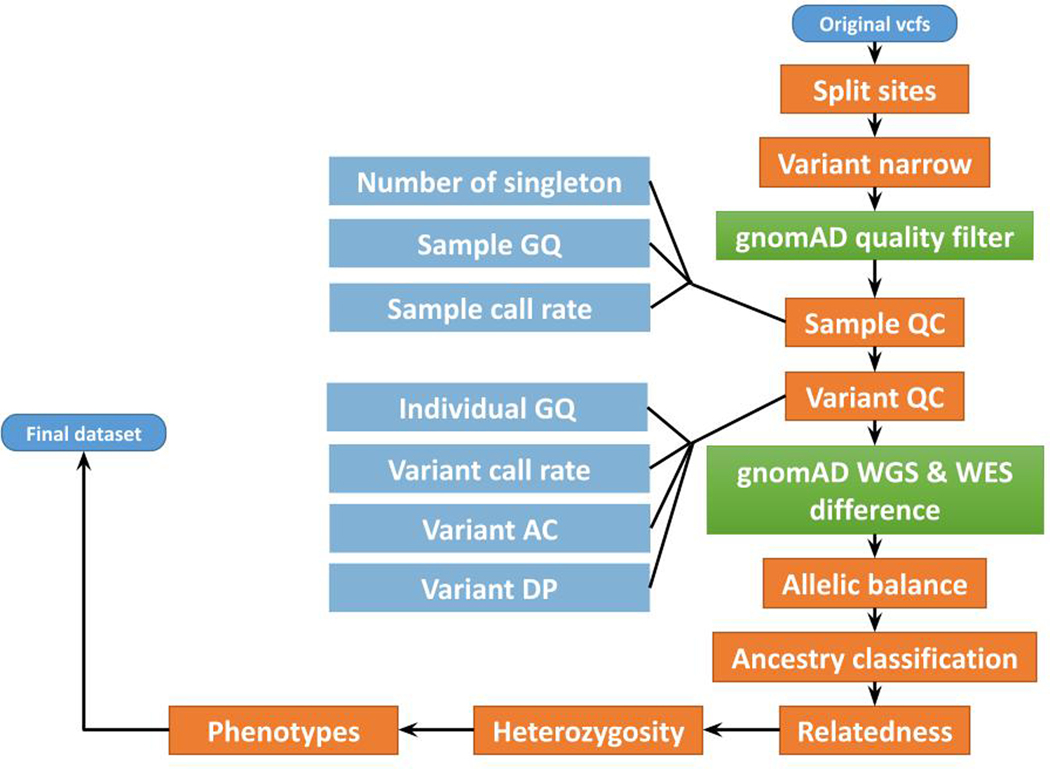
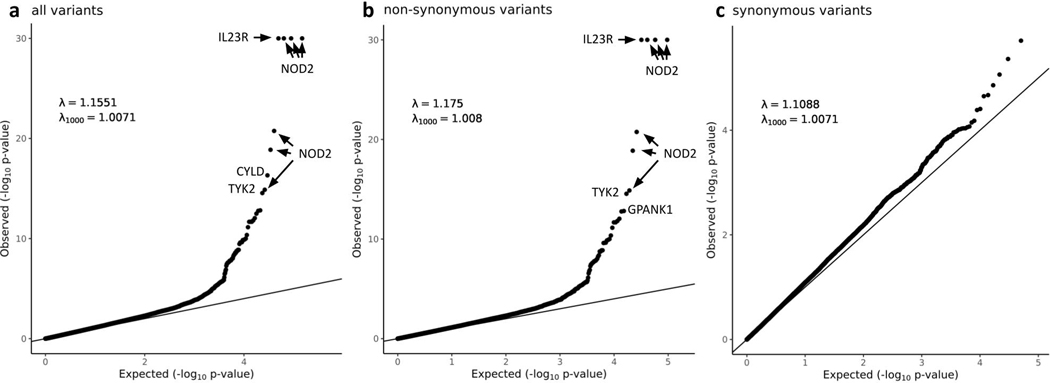
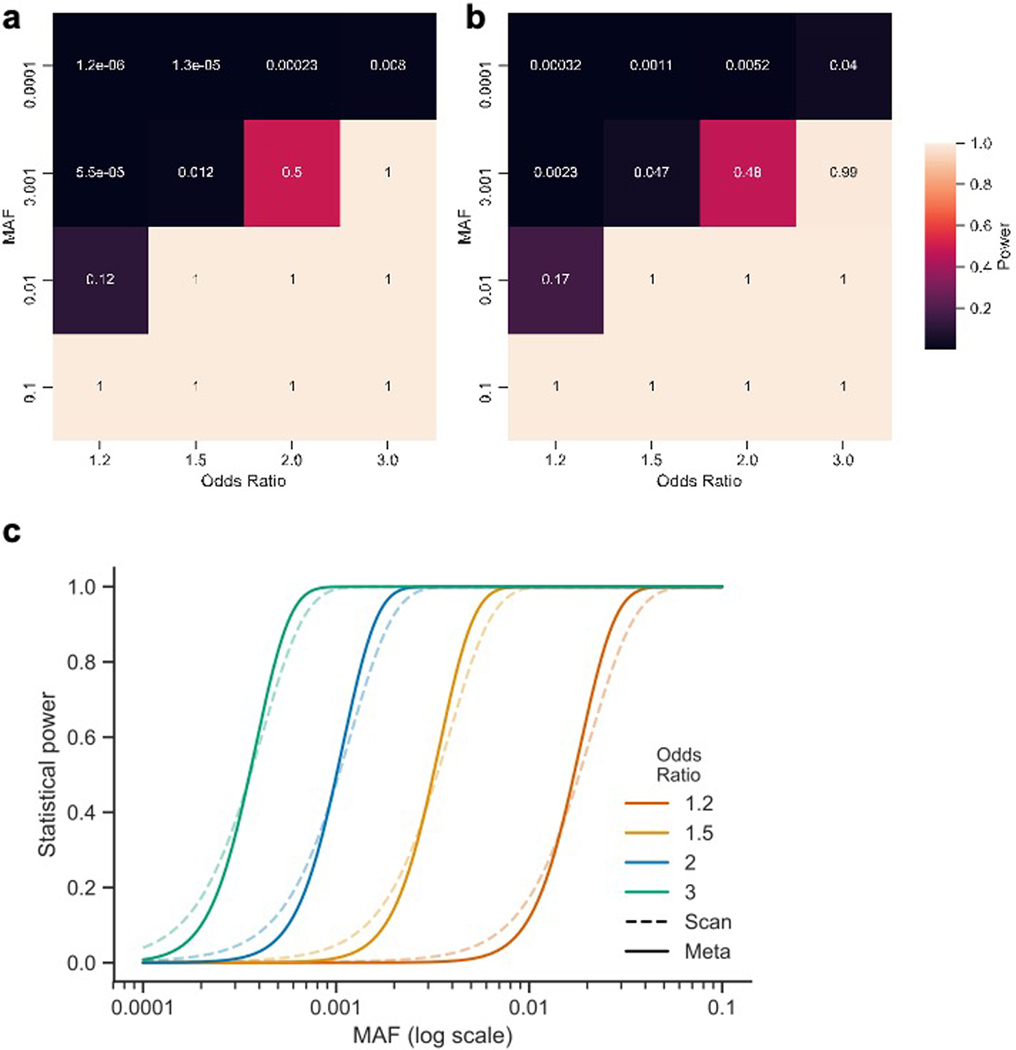
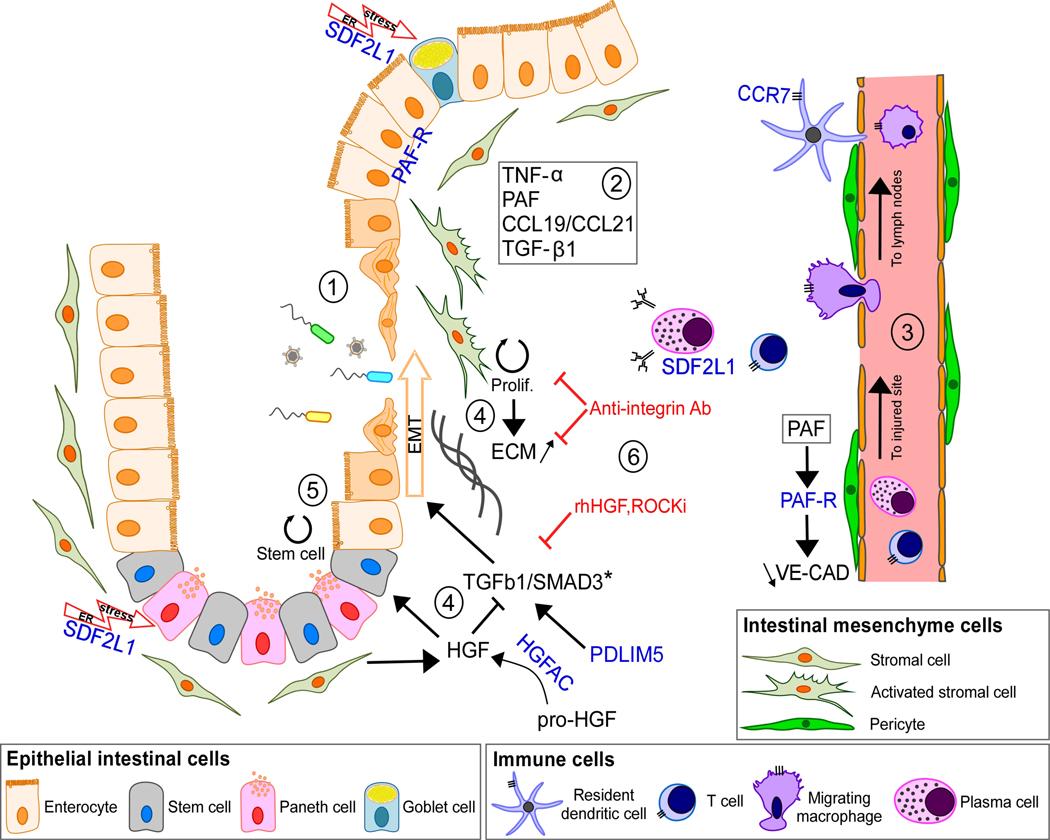
Genome-wide association studies (GWASs) have identified hundreds of loci associated with Crohn's disease (CD). However, as with all complex diseases, robust identification of the genes dysregulated by noncoding variants typically driving GWAS discoveries has been challenging. Here, to complement GWASs and better define actionable biological targets, we analyzed sequence data from more than 30,000 patients with CD and 80,000 population controls. We directly implicate ten genes in general onset CD for the first time to our knowledge via association to coding variation, four of which lie within established CD GWAS loci. In nine instances, a single coding variant is significantly associated, and in the tenth, ATG4C, we see additionally a significantly increased burden of very rare coding variants in CD cases. In addition to reiterating the central role of innate and adaptive immune cells as well as autophagy in CD pathogenesis, these newly associated genes highlight the emerging role of mesenchymal cells in the development and maintenance of intestinal inflammation.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing Interests Statement
A.B., M.F, J.E.H., D.S are current or former employees and/or stockholders of Regeneron Genetics Center or Regeneron Pharmaceuticals. M.A. is consulting for or part of the advisory board for AbbVie Inc, Bellatrix Pharmaceuticals, Bristol Myers Squibb, Eli Lilly Pharmaceuticals, Gilead, Janssen Ortho, LLC, and Prometheus Biosciences; teaching, lecturing, or speaking at Alimentiv, Arena Pharmaceuticals, Janssen, Prime CME, Takeda Pharmaceuticals. A.B is an employee of Regeneron and owns stock in Regeneron. O.M.D. has served in the IBD fellowship funding committee for Pfizer and has a funded research project by Pfizer. H.K. receives grant funding from Takeda and Pfizer and has received consulting fees from Takeda. A.P. is a member of Astra Zenecas Genomics Advisory Board. M.A.R. is on the SAB of 54gene and has advised BioMarin, Third Rock Ventures, MazeTx, and Related Sciences. G.A.H. is an employee of Takeda, former employee of AbbVie and owns stock in Takeda and AbbVie. C.A.L. reports grants from Genentech, grants and personal fees from Janssen, grants and personal fees from Takeda, grants from AbbVie, personal fees from Ferring, grants from Eli Lilly, grants from Pfizer, grants from Roche, grants from UCB Biopharma, grants from Sanofi Aventis, grants from Biogen IDEC, grants from Orion OYJ, personal fees from Dr Falk Pharma, grants from AstraZeneca, outside the submitted work. H.H.U reports research collaboration or consultancy with Janssen, Eli Lilly, UCB Pharma, Celgene, MiroBio, OMass, and Mestag. D.P.B.M. has consulted for Takeda, Boehringer Ingelheim, Palatin Technologies, Bridge Biotherapeutics, Pfizer, and Gilead. M.P. received an unrestricted research grant from Pfizer UK and speaker fees from Janssen. P.I. received lecture fees from AbbVie, BMS, Celgene, Celltrion, Falk Pharma, Ferring, Galapagos, Gilead, MSD, Janssen, Pfizer, Takeda, Tillotts, Sapphire Medical, Sandoz, Shire and Warner Chilcott; financial support for research from Celltrion, MSD, Pfizer and Takeda; advisory fees from AbbVie, Arena, Boehringer-Ingelheim, BMS, Celgene, Celltrion, Genentech, Gilead, Hospira, Janssen, Lilly, MSD, Pfizer, Pharmacosmos, Prometheus, Roche, Sandoz, Samsung Bioepis, Takeda, Topivert, VH2, Vifor Pharma and Warner Chilcott. Cedars-Sinai and D.P.B.M. have financial interests in Prometheus Biosciences, a company which has access to the data and specimens in Cedars-Sinais MIRIAD Biobank (including the Cedars-Sinai data and specimens used in this study) and seeks to develop commercial products. H.H. has received consultancy fees from Ono Pharmaceutical and honorarium from Xian Janssen Pharmaceutical. C.A.A. has received consultancy fees from Genomics plc and BridgeBio Inc. and lecture fees from GSK. M.J.D. is a founder of Maze Therapeutics. The remaining authors declare no competing interests.
Figures
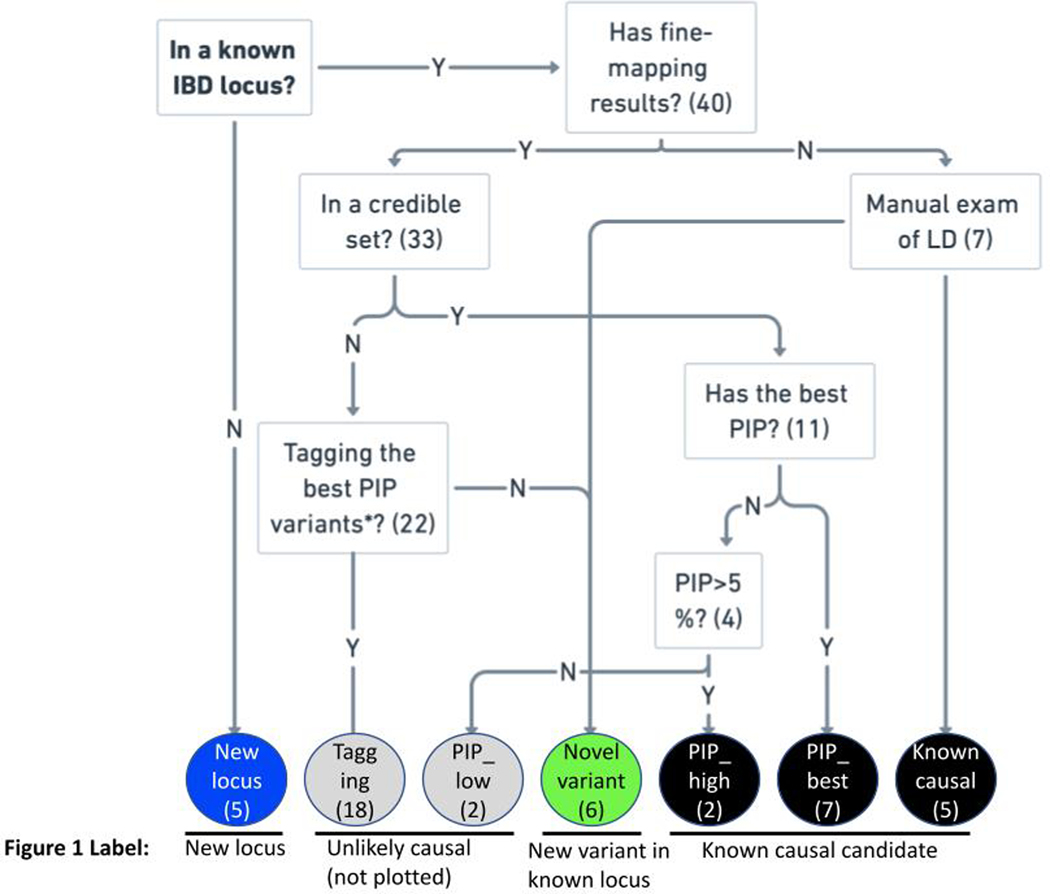
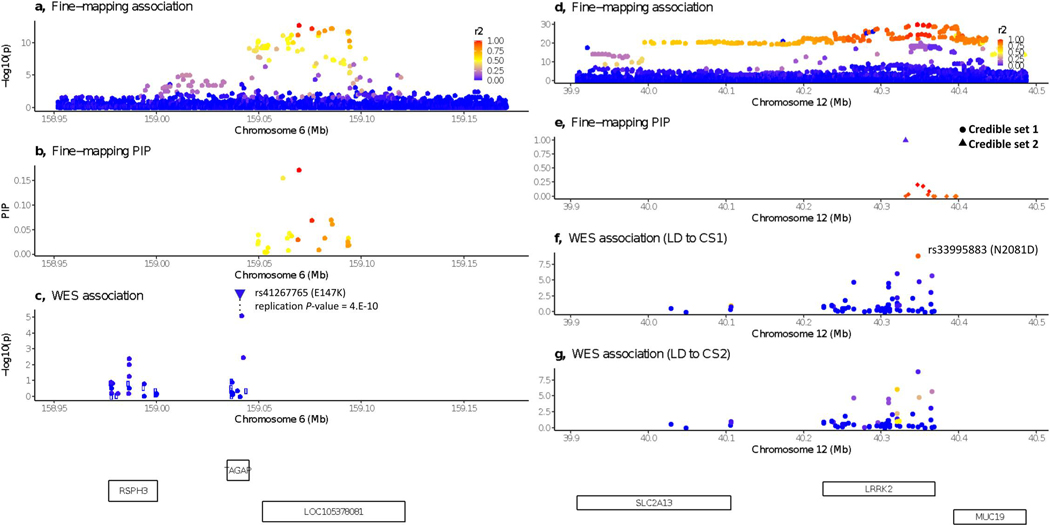
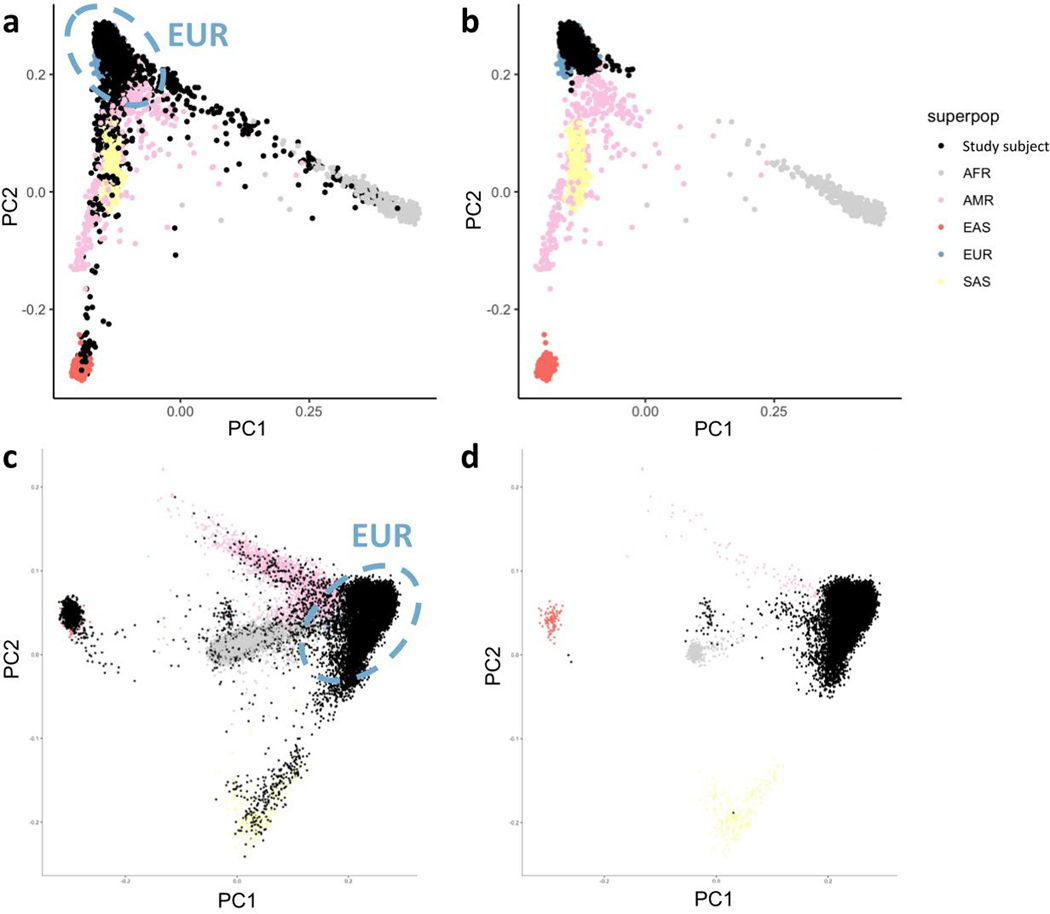
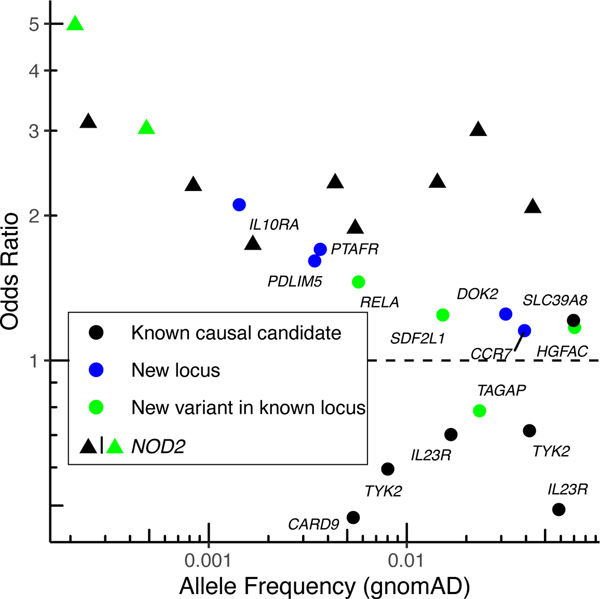
References
Publication types
MeSH terms
Grants and funding
- P01 DK094779/DK/NIDDK NIH HHS/United States
- 206194 /WT_/Wellcome Trust/United Kingdom
- K01 DK114379/DK/NIDDK NIH HHS/United States
- MR/M00533X/1/MRC_/Medical Research Council/United Kingdom
- R01 HG010140/HG/NHGRI NIH HHS/United States
- U01 DK062423/DK/NIDDK NIH HHS/United States
- MC_UU_00008/7/MRC_/Medical Research Council/United Kingdom
- MC_UU_12010/7/MRC_/Medical Research Council/United Kingdom
- U54 HG003067/HG/NHGRI NIH HHS/United States
- U01 DK062413/DK/NIDDK NIH HHS/United States
- UM1 HG008895/HG/NHGRI NIH HHS/United States
- P30 DK043351/DK/NIDDK NIH HHS/United States
- DH_/Department of Health/United Kingdom
- U01 DK062432/DK/NIDDK NIH HHS/United States
- R01 DK104844/DK/NIDDK NIH HHS/United States
- MC_PC_MR/S025952/1/MRC_/Medical Research Council/United Kingdom
- 108413/A/15/D/WT_/Wellcome Trust/United Kingdom
- U01 HG009080/HG/NHGRI NIH HHS/United States
- K23 DK117054/DK/NIDDK NIH HHS/United States
- R01 DK111843/DK/NIDDK NIH HHS/United States
- P30 AG038072/AG/NIA NIH HHS/United States
- R01 DK127044/DK/NIDDK NIH HHS/United States
- MR/S036377/1/MRC_/Medical Research Council/United Kingdom
- P01 DK046763/DK/NIDDK NIH HHS/United States
- P30 DK052574/DK/NIDDK NIH HHS/United States
- U01 DK062420/DK/NIDDK NIH HHS/United States
- U01 DK062431/DK/NIDDK NIH HHS/United States
- R01 DK087694/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
