

Multiple genome alignment in the telomere-to-telomere assembly era
- PMID: 36038949
- PMCID: PMC9421119
- DOI: 10.1186/s13059-022-02735-6
Multiple genome alignment in the telomere-to-telomere assembly era
Abstract
With the arrival of telomere-to-telomere (T2T) assemblies of the human genome comes the computational challenge of efficiently and accurately constructing multiple genome alignments at an unprecedented scale. By identifying nucleotides across genomes which share a common ancestor, multiple genome alignments commonly serve as the bedrock for comparative genomics studies. In this review, we provide an overview of the algorithmic template that most multiple genome alignment methods follow. We also discuss prospective areas of improvement of multiple genome alignment for keeping up with continuously arriving high-quality T2T assembled genomes and for unlocking clinically-relevant insights.
Keywords: Comparative genomics; Homology; Multiple genome alignment; Synteny.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
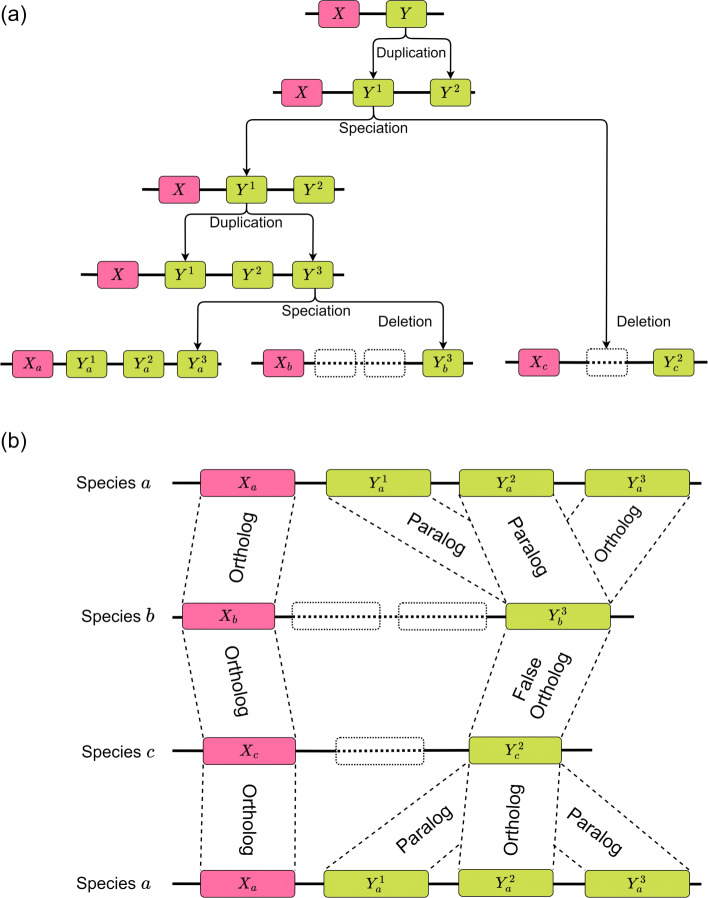
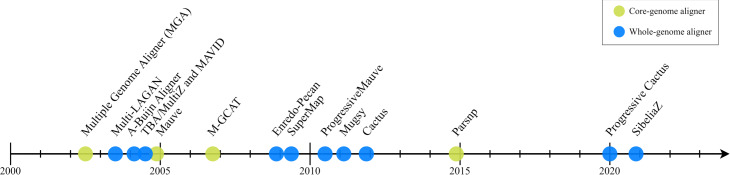
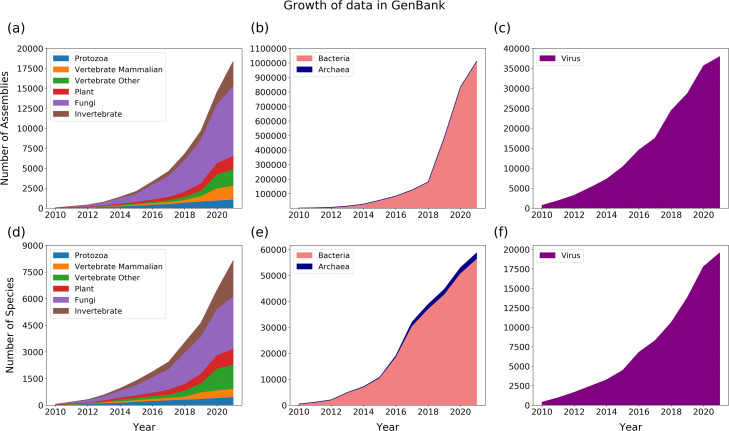
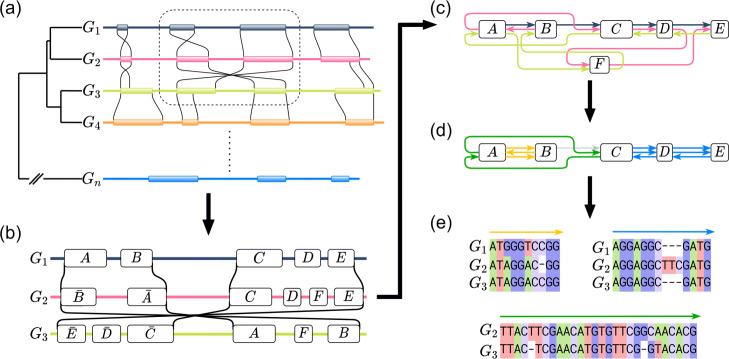
Similar articles
-
Unlocking plant genetics with telomere-to-telomere genome assemblies.Nat Genet. 2024 Sep;56(9):1788-1799. doi: 10.1038/s41588-024-01830-7. Epub 2024 Jul 24. Nat Genet. 2024. PMID: 39048791 Review.
-
Structural variation in humans and our primate kin in the era of telomere-to-telomere genomes and pangenomics.Curr Opin Genet Dev. 2024 Aug;87:102233. doi: 10.1016/j.gde.2024.102233. Epub 2024 Jul 23. Curr Opin Genet Dev. 2024. PMID: 39042999 Free PMC article. Review.
-
T2T-YAO: A Telomere-to-telomere Assembled Diploid Reference Genome for Han Chinese.Genomics Proteomics Bioinformatics. 2023 Dec;21(6):1085-1100. doi: 10.1016/j.gpb.2023.08.001. Epub 2023 Aug 16. Genomics Proteomics Bioinformatics. 2023. PMID: 37595788 Free PMC article.
-
Localizing unmapped sequences with families to validate the Telomere-to-Telomere assembly and identify new hotspots for genetic diversity.Genome Res. 2023 Oct;33(10):1734-1746. doi: 10.1101/gr.277175.122. Epub 2023 Oct 25. Genome Res. 2023. PMID: 37879860 Free PMC article.
-
Envisioning a new era: Complete genetic information from routine, telomere-to-telomere genomes.Am J Hum Genet. 2023 Nov 2;110(11):1832-1840. doi: 10.1016/j.ajhg.2023.09.011. Am J Hum Genet. 2023. PMID: 37922882 Free PMC article. Review.
Cited by
-
EvANI benchmarking workflow for evolutionary distance estimation.bioRxiv [Preprint]. 2025 Feb 23:2025.02.23.639716. doi: 10.1101/2025.02.23.639716. bioRxiv. 2025. Update in: Brief Bioinform. 2025 May 1;26(3):bbaf267. doi: 10.1093/bib/bbaf267. PMID: 40027788 Free PMC article. Updated. Preprint.
-
EvANI benchmarking workflow for evolutionary distance estimation.Brief Bioinform. 2025 May 1;26(3):bbaf267. doi: 10.1093/bib/bbaf267. Brief Bioinform. 2025. PMID: 40501070 Free PMC article.
-
Pan-conserved segment tags identify ultra-conserved sequences across assemblies in the human pangenome.Cell Rep Methods. 2023 Aug 2;3(8):100543. doi: 10.1016/j.crmeth.2023.100543. eCollection 2023 Aug 28. Cell Rep Methods. 2023. PMID: 37671027 Free PMC article.
-
ACMGA: a reference-free multiple-genome alignment pipeline for plant species.BMC Genomics. 2024 May 25;25(1):515. doi: 10.1186/s12864-024-10430-y. BMC Genomics. 2024. PMID: 38796435 Free PMC article.
-
Telomere-to-telomere gap-free genome assembly provides genetic insight into the triterpenoid saponins biosynthesis in Platycodon grandiflorus.Hortic Res. 2025 Feb 1;12(5):uhaf030. doi: 10.1093/hr/uhaf030. eCollection 2025 May. Hortic Res. 2025. PMID: 40224331 Free PMC article.
References
-
- Hannenhalli S, Pevzner PA. Proceedings of IEEE 36th Annual Foundations of Computer Science. New York: IEEE; 1995. Transforming men into mice (polynomial algorithm for genomic distance problem)
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
