

Accurate de novo design of membrane-traversing macrocycles
- PMID: 36041435
- PMCID: PMC9490236
- DOI: 10.1016/j.cell.2022.07.019
Accurate de novo design of membrane-traversing macrocycles
Abstract
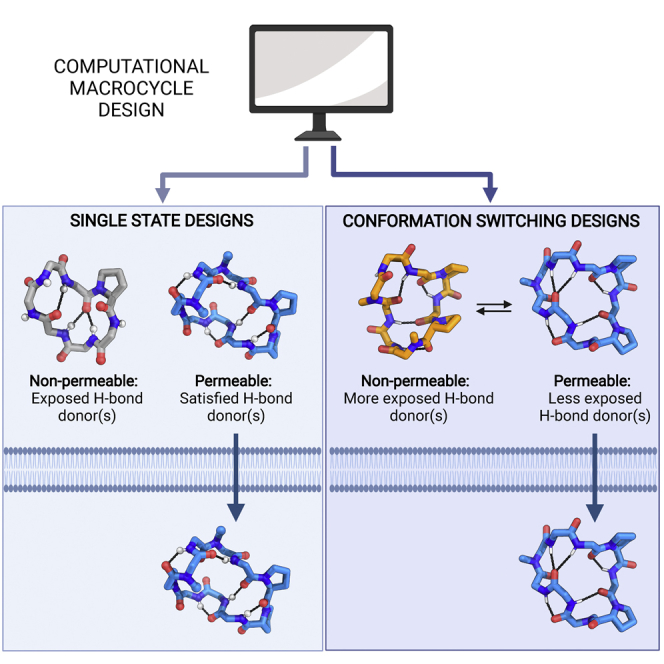
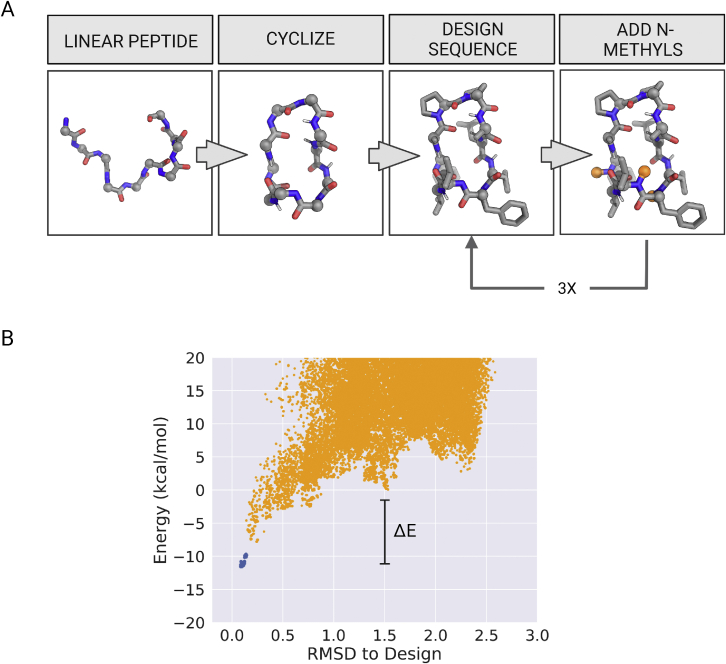
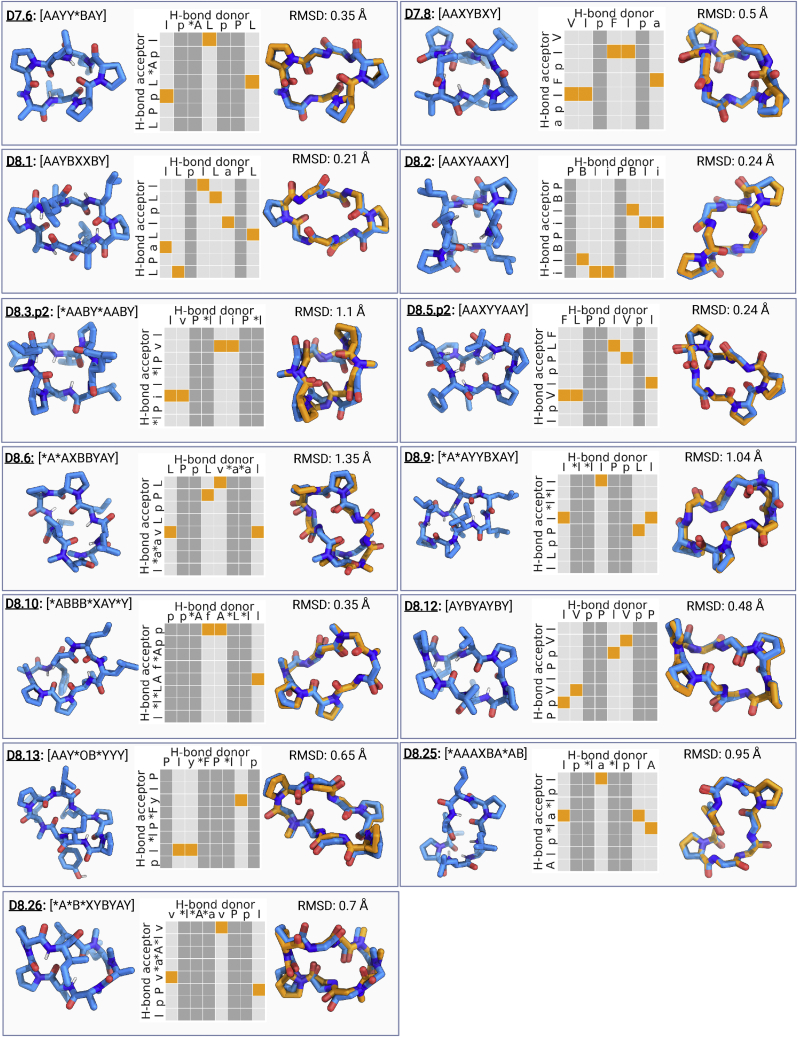
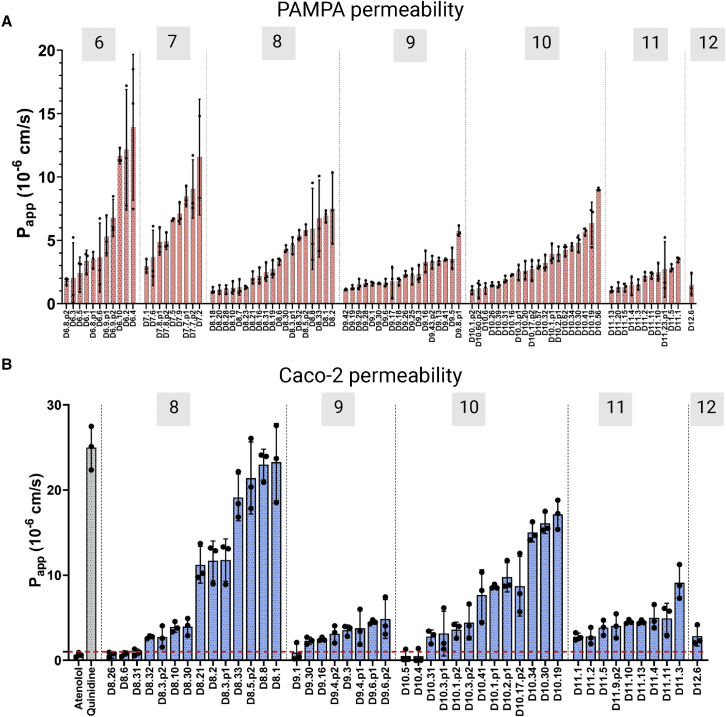
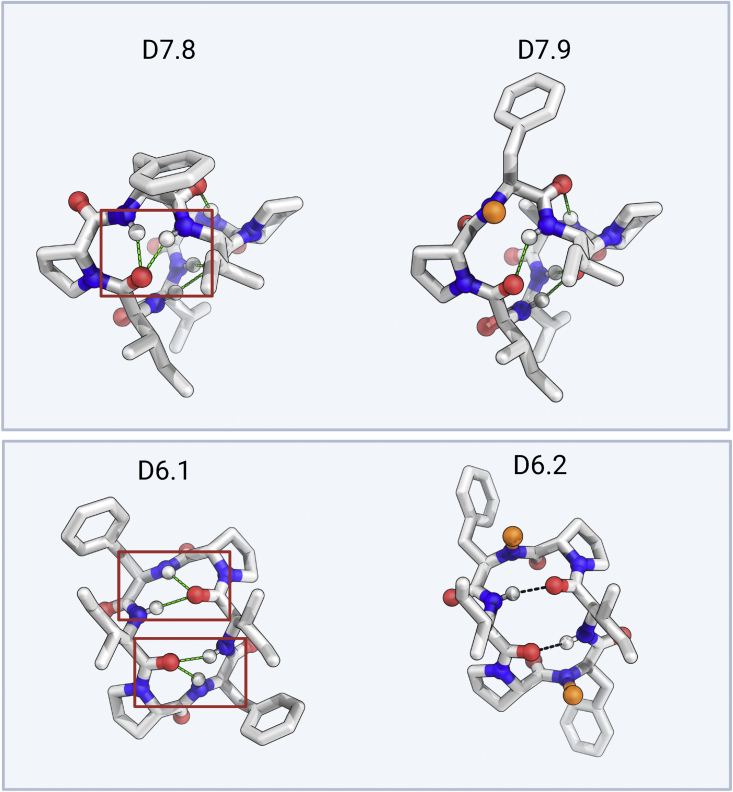
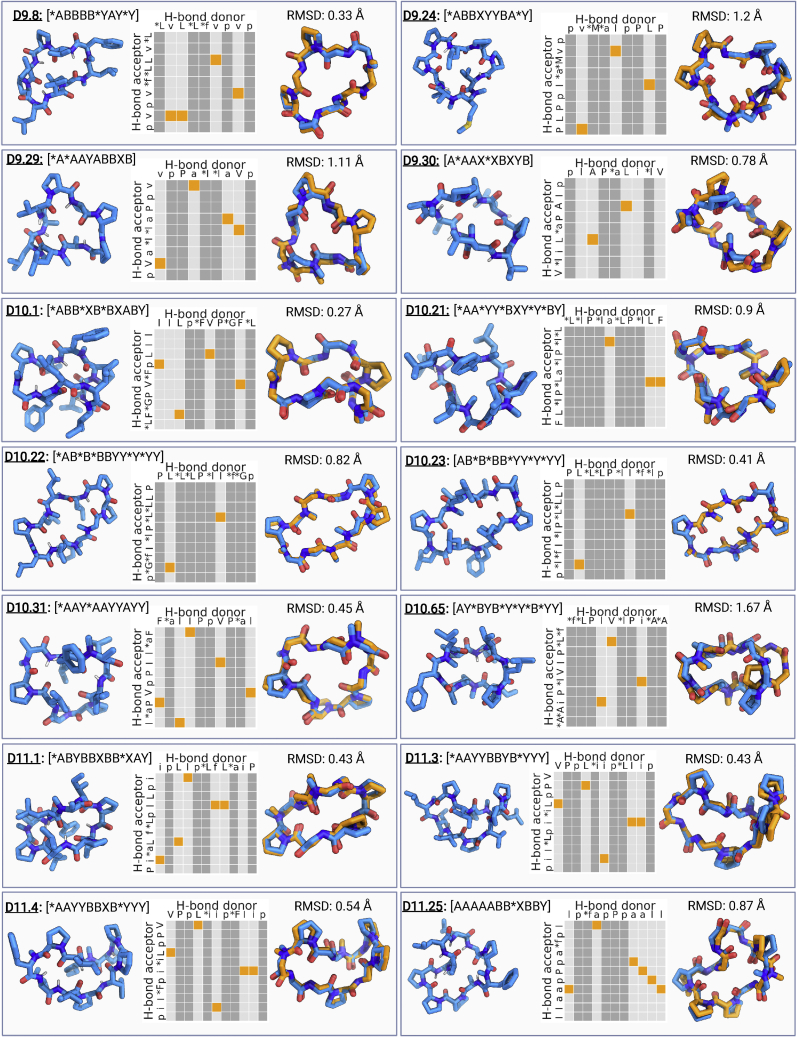
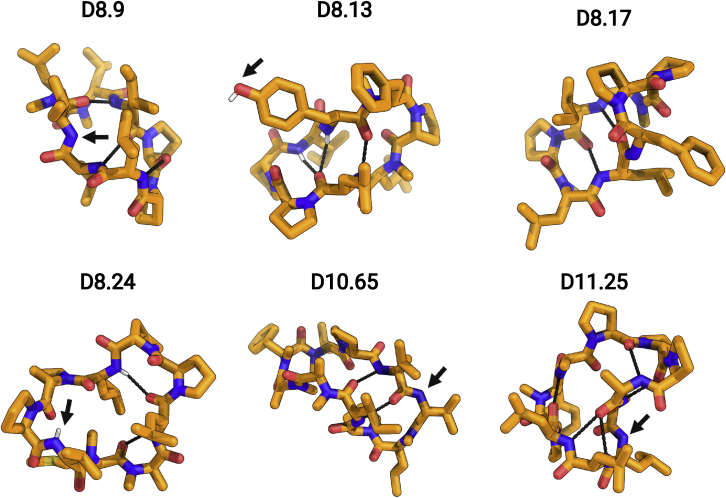
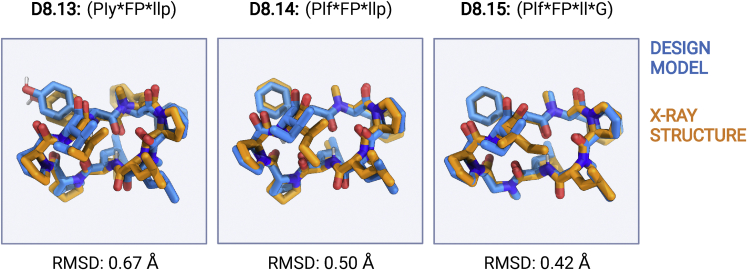
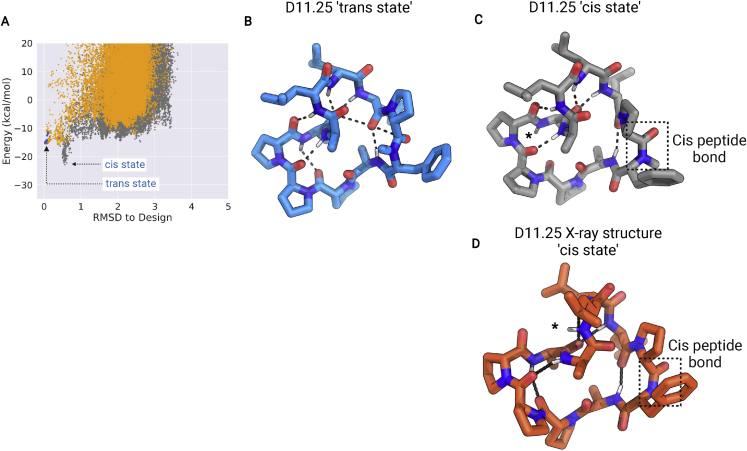
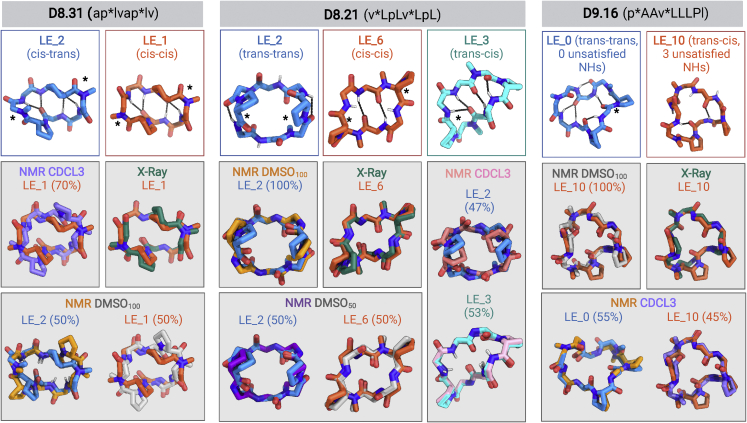
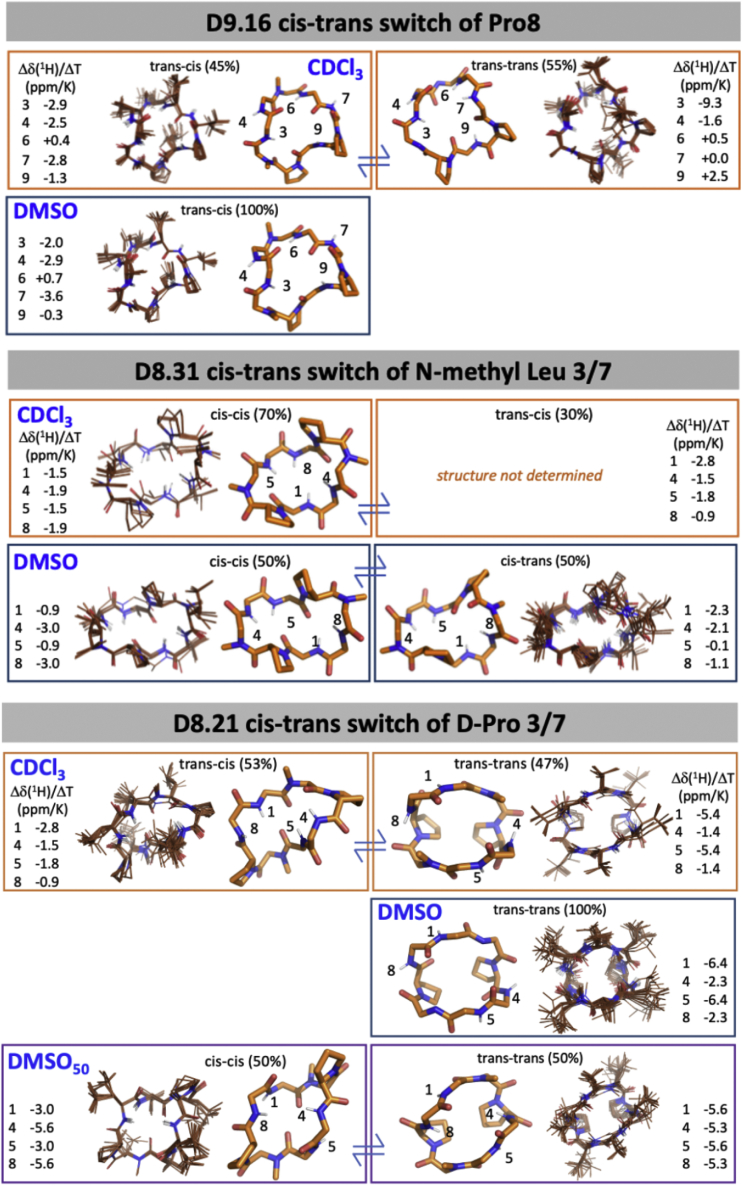
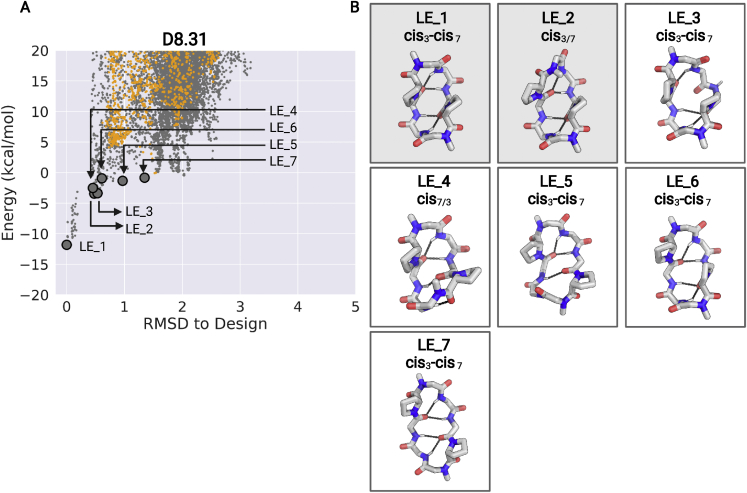
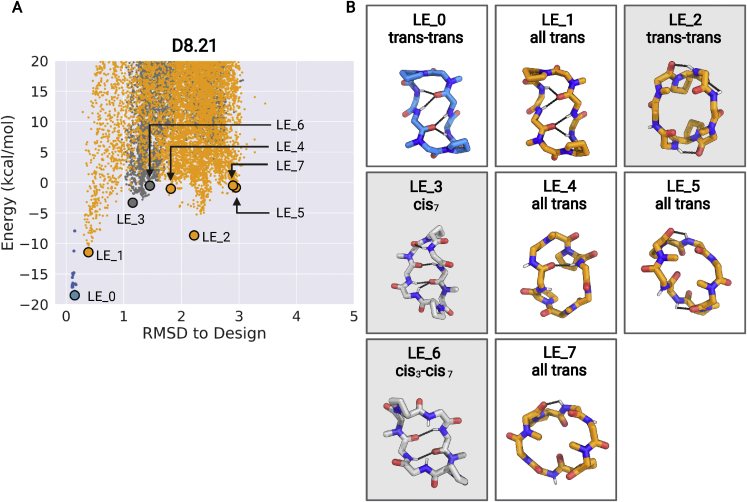
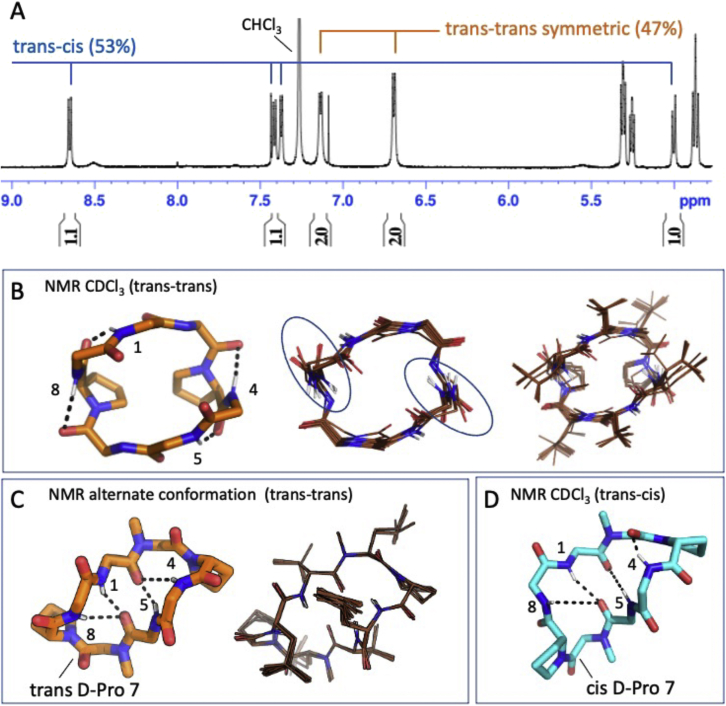
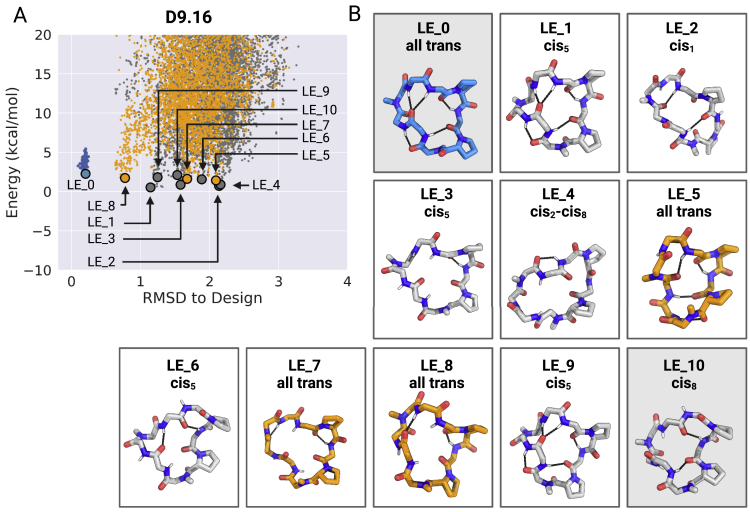
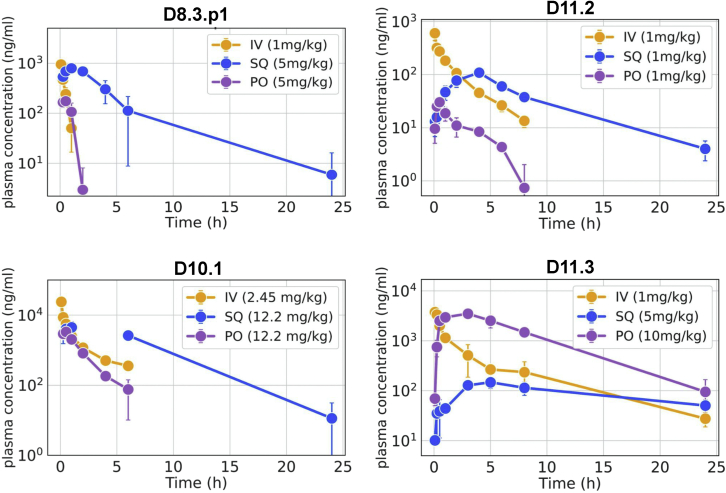
We use computational design coupled with experimental characterization to systematically investigate the design principles for macrocycle membrane permeability and oral bioavailability. We designed 184 6-12 residue macrocycles with a wide range of predicted structures containing noncanonical backbone modifications and experimentally determined structures of 35; 29 are very close to the computational models. With such control, we show that membrane permeability can be systematically achieved by ensuring all amide (NH) groups are engaged in internal hydrogen bonding interactions. 84 designs over the 6-12 residue size range cross membranes with an apparent permeability greater than 1 × 10-6 cm/s. Designs with exposed NH groups can be made membrane permeable through the design of an alternative isoenergetic fully hydrogen-bonded state favored in the lipid membrane. The ability to robustly design membrane-permeable and orally bioavailable peptides with high structural accuracy should contribute to the next generation of designed macrocycle therapeutics.
Keywords: computational design; membrane permeability; oral bioavailability; peptide design.
Copyright © 2022 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests V.K.M. is a cofounder and shareholder of Menten AI, a biotechnology company. G.T.M. is a cofounder of Nexomics Biosciences, Inc., a structural biology contract research organization. A provisional patent covering the membrane-permeable peptides described in this paper has been filed by the University of Washington, Seattle. G.B., L.S., and D.B. are cofounders and shareholders of an early-stage biotechnology company that has licensed the provisional patent.
Figures
Comment in
-
Predicting permeable macrocycles.Nat Rev Drug Discov. 2022 Nov;21(11):798. doi: 10.1038/d41573-022-00166-3. Nat Rev Drug Discov. 2022. PMID: 36192644 No abstract available.
References
-
- Baxter N.J., Williamson M.P. Temperature dependence of 1H chemical shifts in proteins. J. Biomol. NMR. 1997;9:359–369. - PubMed
-
- Bockus A.T., Lexa K.W., Pye C.R., Kalgutkar A.S., Gardner J.W., Hund K.C.R., Hewitt W.M., Schwochert J.A., Glassey E., Price D.A., et al. Probing the physicochemical boundaries of cell permeability and oral bioavailability in lipophilic macrocycles inspired by natural products. J. Med. Chem. 2015;58:4581–4589. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
