

Engineering functional thermostable proteins using ancestral sequence reconstruction
- PMID: 36041629
- PMCID: PMC9525910
- DOI: 10.1016/j.jbc.2022.102435
Engineering functional thermostable proteins using ancestral sequence reconstruction
Abstract
Natural proteins are often only slightly more stable in the native state than the denatured state, and an increase in environmental temperature can easily shift the balance toward unfolding. Therefore, the engineering of proteins to improve protein stability is an area of intensive research. Thermostable proteins are required to withstand industrial process conditions, for increased shelf-life of protein therapeutics, for developing robust 'biobricks' for synthetic biology applications, and for research purposes (e.g., structure determination). In addition, thermostability buffers the often destabilizing effects of mutations introduced to improve other properties. Rational design approaches to engineering thermostability require structural information, but even with advanced computational methods, it is challenging to predict or parameterize all the relevant structural factors with sufficient precision to anticipate the results of a given mutation. Directed evolution is an alternative when structures are unavailable but requires extensive screening of mutant libraries. Recently, however, bioinspired approaches based on phylogenetic analyses have shown great promise. Leveraging the rapid expansion in sequence data and bioinformatic tools, ancestral sequence reconstruction can generate highly stable folds for novel applications in industrial chemistry, medicine, and synthetic biology. This review provides an overview of the factors important for successful inference of thermostable proteins by ancestral sequence reconstruction and what it can reveal about the determinants of stability in proteins.
Keywords: ancestral sequence reconstruction; biocatalysis; cytochrome P450; directed evolution; molecular evolution; precambrian; protein engineering; synthetic biology; thermostability.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors are engaged in directed evolution efforts to produce thermostable cytochrome P450 enzymes for biocatalysis and synthetic biology applications, some of which have been licensed for application in pharmaceutical and fine chemical production under the tradename “CYPerior.” The authors declare that they have no conflicts of interest with the contents of this article.
Figures

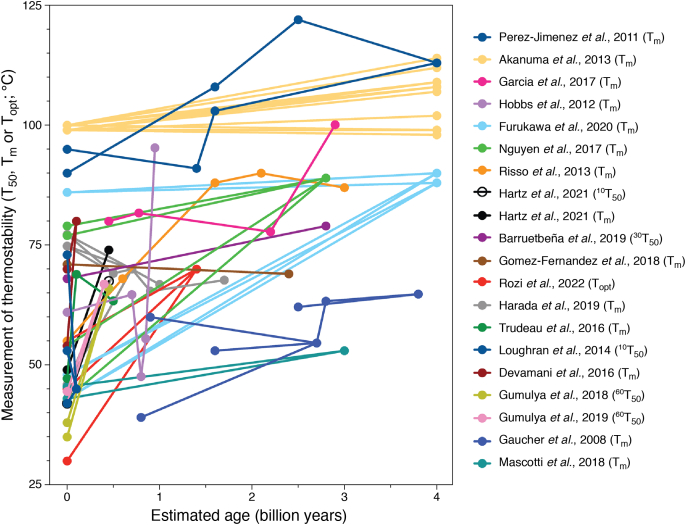
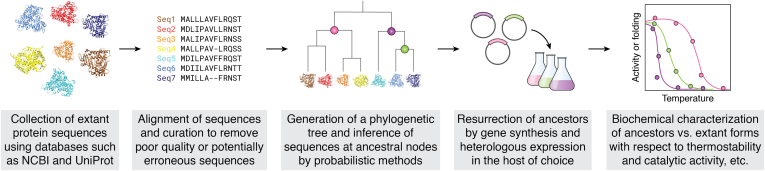
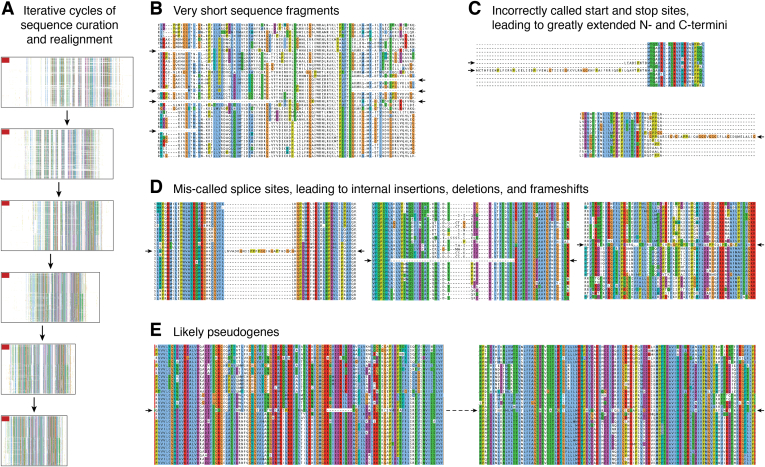
References
-
- Pace C.N. Conformational stability of globular proteins. Trends Biochem. Sci. 1990;15:14–17. - PubMed
-
- Bommarius A.S., Paye M.F. Stabilizing biocatalysts. Chem. Soc. Rev. 2013;42:6534–6565. - PubMed
-
- Burton S.G., Cowan D.A., Woodley J.M. The search for the ideal biocatalyst. Nat. Biotech. 2002;20:37–45. - PubMed
-
- Tokuriki N., Tawfik D.S. Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 2009;19:596–604. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
