

Mitochondrial DNA is a major source of driver mutations in cancer
- PMID: 36041967
- PMCID: PMC9671861
- DOI: 10.1016/j.trecan.2022.08.001
Mitochondrial DNA is a major source of driver mutations in cancer
Abstract
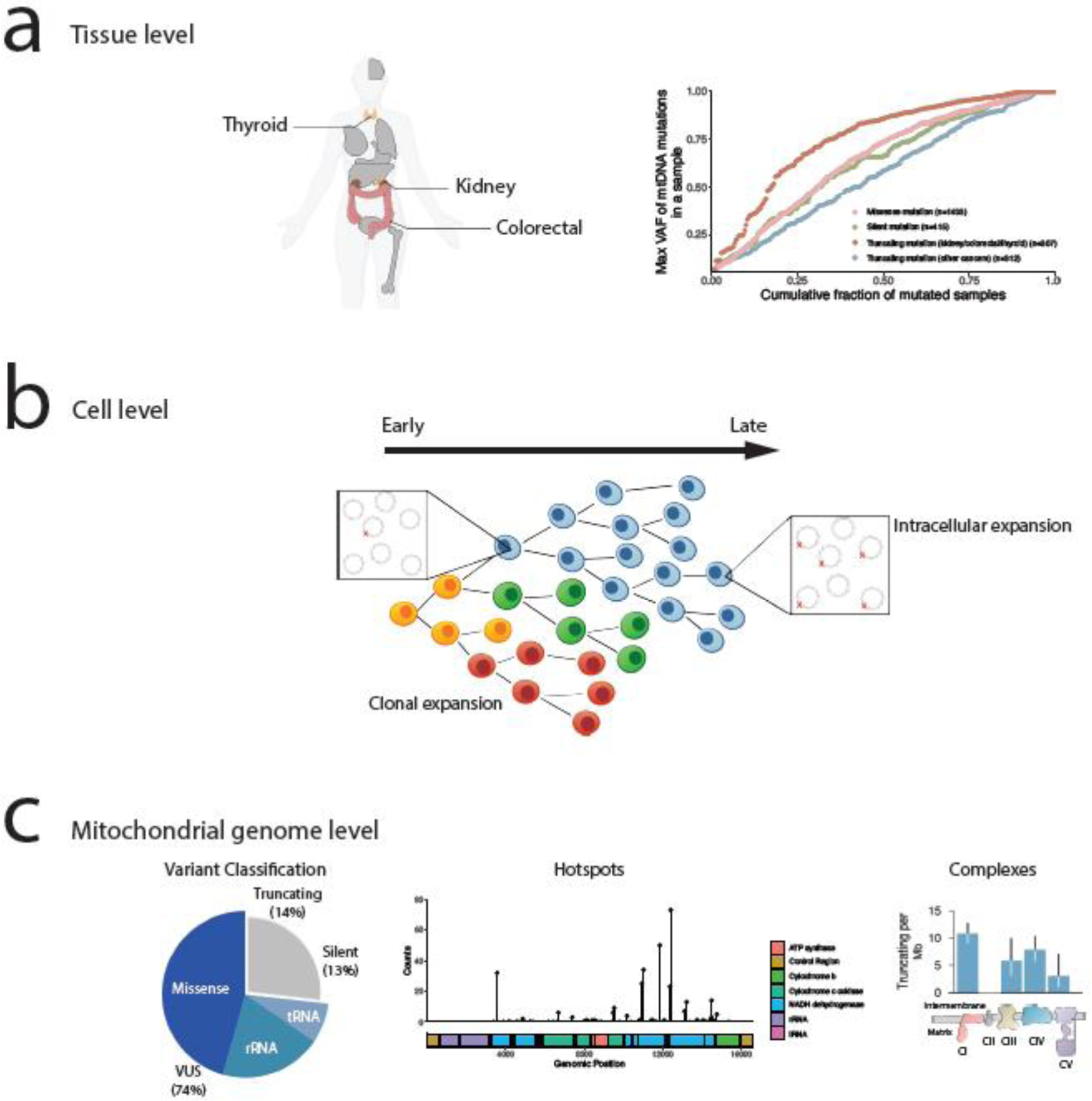
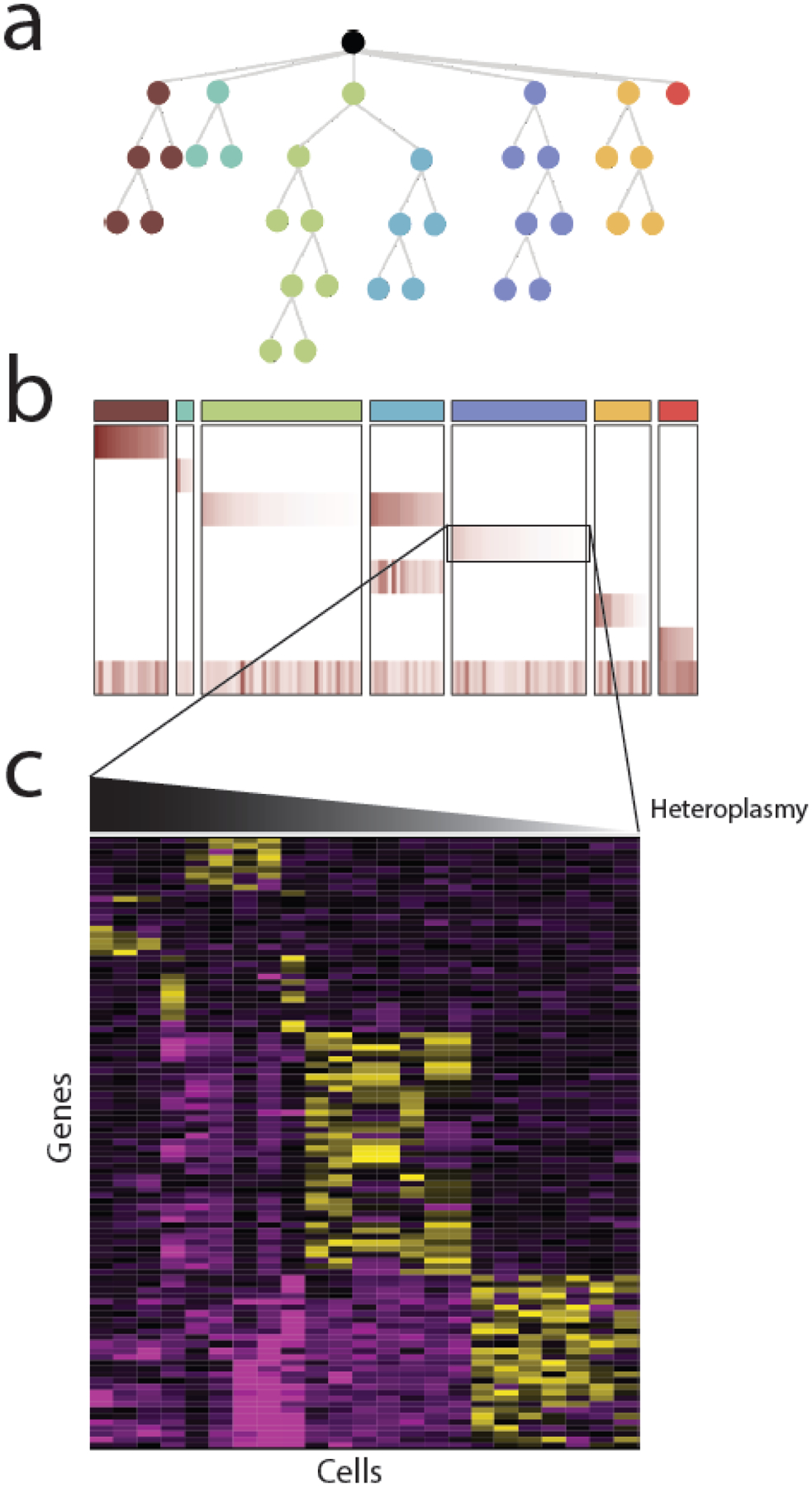
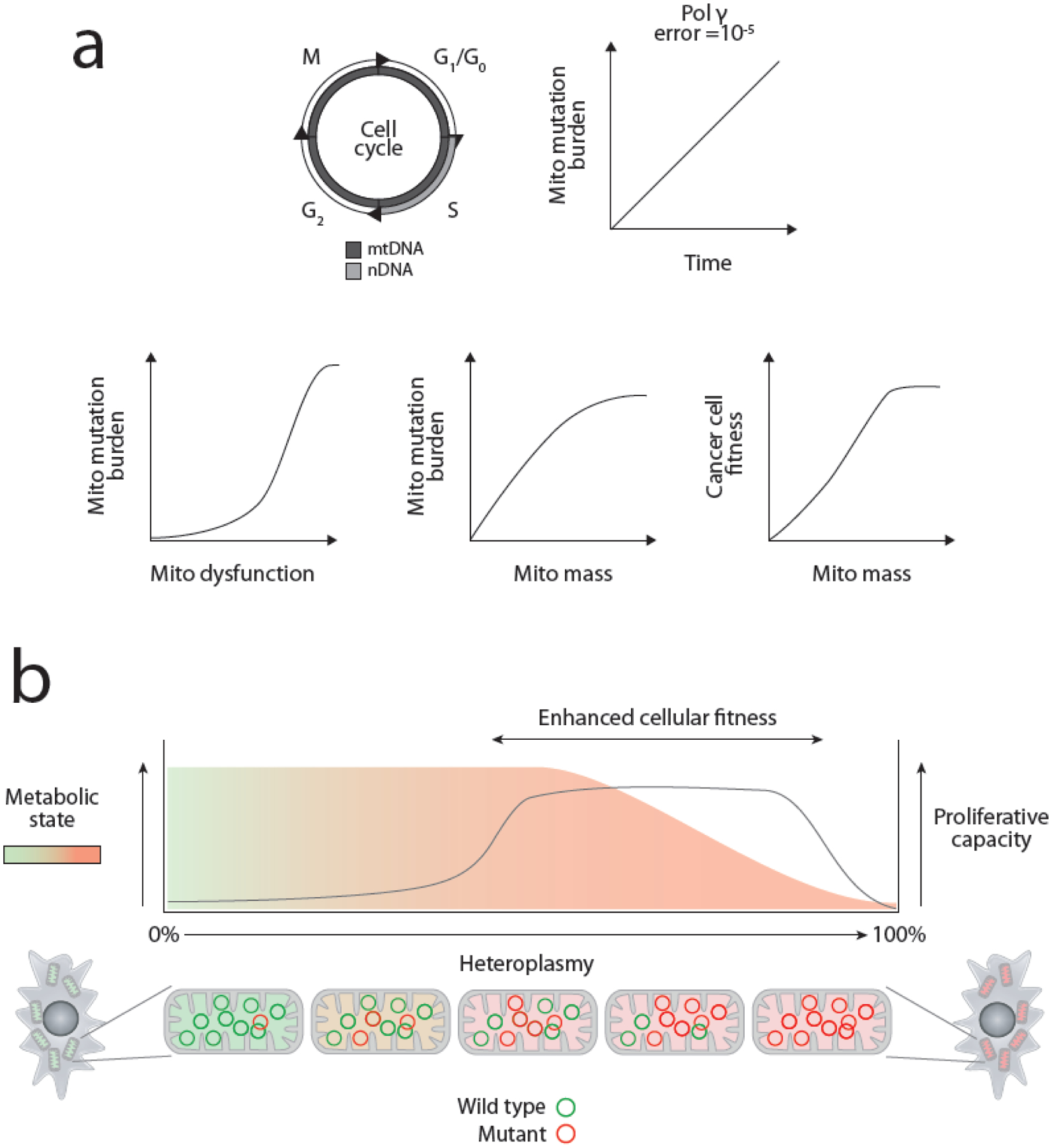
Mitochondrial DNA (mtDNA) mutations are among the most common genetic events in all tumors and directly impact metabolic homeostasis. Despite the central role mitochondria play in energy metabolism and cellular physiology, the role of mutations in the mitochondrial genomes of tumors has been contentious. Until recently, genomic and functional studies of mtDNA variants were impeded by a lack of adequate tumor mtDNA sequencing data and available methods for mitochondrial genome engineering. These barriers and a conceptual fog surrounding the functional impact of mtDNA mutations in tumors have begun to lift, revealing a path to understanding the role of this essential metabolic genome in cancer initiation and progression. Here we discuss the history, recent developments, and challenges that remain for mitochondrial oncogenetics as the impact of a major new class of cancer-associated mutations is unveiled.
Keywords: cancer; genome editing; mitochondrial DNA; mutation selection.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
