

Controlling gene expression with deep generative design of regulatory DNA
- PMID: 36042233
- PMCID: PMC9427793
- DOI: 10.1038/s41467-022-32818-8
Controlling gene expression with deep generative design of regulatory DNA
Abstract
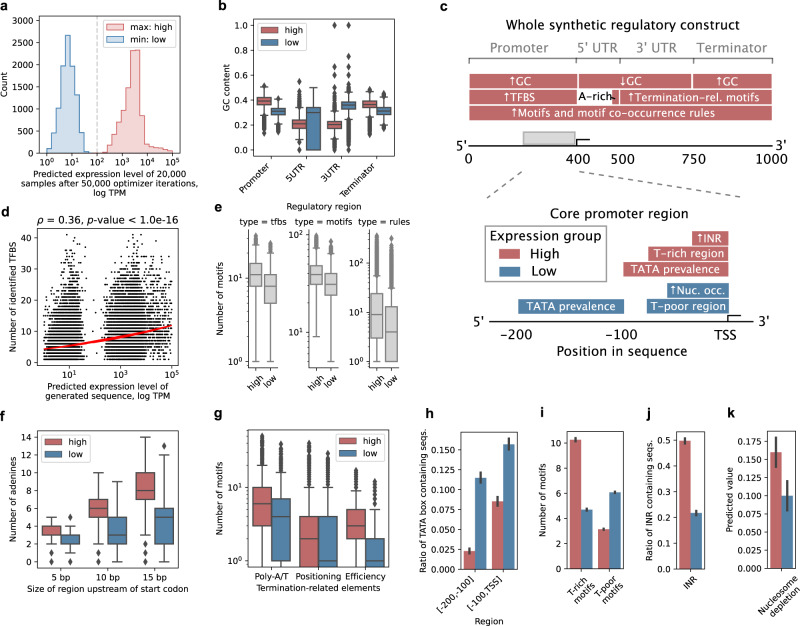
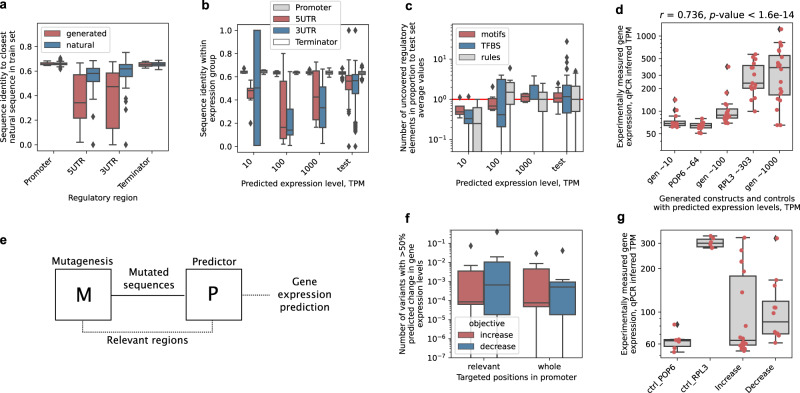
Design of de novo synthetic regulatory DNA is a promising avenue to control gene expression in biotechnology and medicine. Using mutagenesis typically requires screening sizable random DNA libraries, which limits the designs to span merely a short section of the promoter and restricts their control of gene expression. Here, we prototype a deep learning strategy based on generative adversarial networks (GAN) by learning directly from genomic and transcriptomic data. Our ExpressionGAN can traverse the entire regulatory sequence-expression landscape in a gene-specific manner, generating regulatory DNA with prespecified target mRNA levels spanning the whole gene regulatory structure including coding and adjacent non-coding regions. Despite high sequence divergence from natural DNA, in vivo measurements show that 57% of the highly-expressed synthetic sequences surpass the expression levels of highly-expressed natural controls. This demonstrates the applicability and relevance of deep generative design to expand our knowledge and control of gene expression regulation in any desired organism, condition or tissue.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
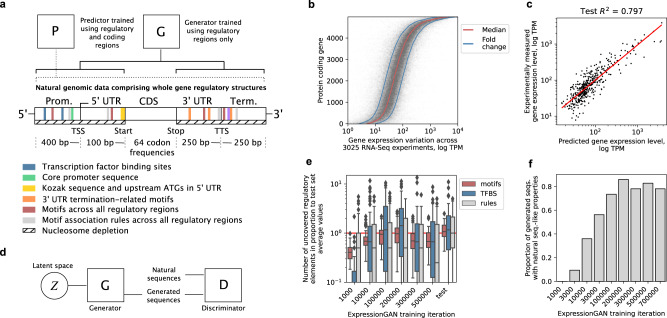
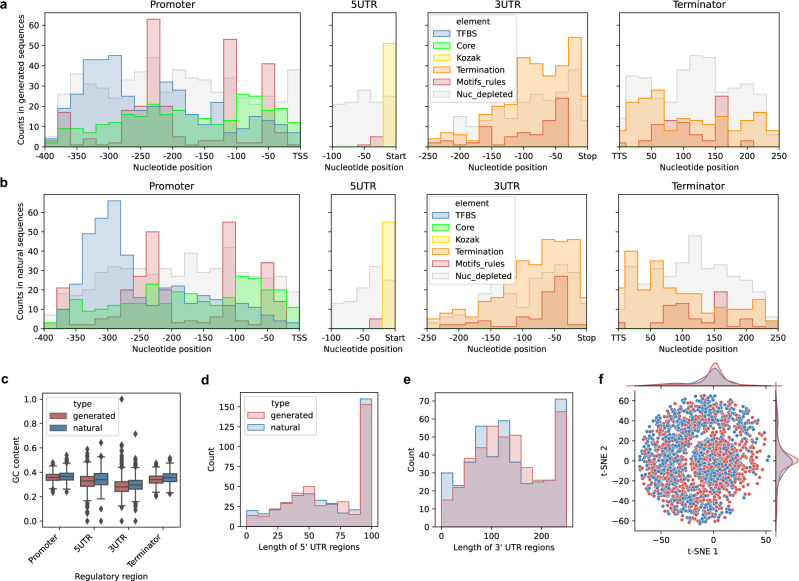
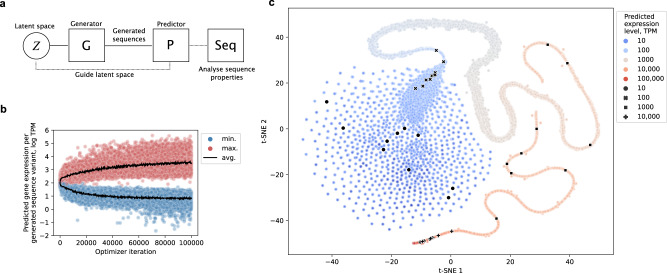
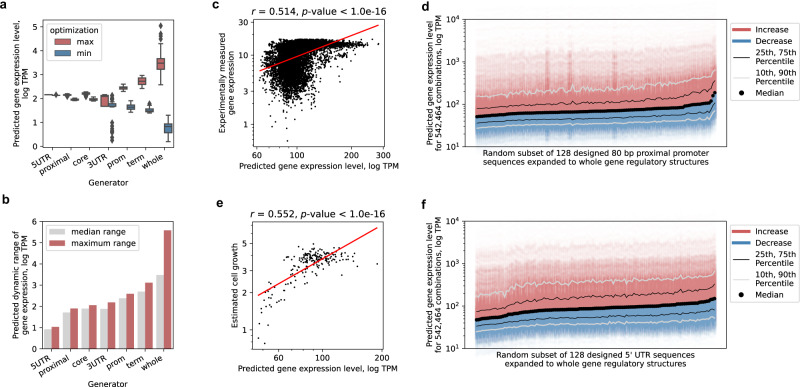
References
-
- Dunbar, C. E. et al. Gene therapy comes of age. Science359, eaan4672 (2018). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
