

Genomic diversity and biosynthetic capabilities of sponge-associated chlamydiae
- PMID: 36042324
- PMCID: PMC9666466
- DOI: 10.1038/s41396-022-01305-9
Genomic diversity and biosynthetic capabilities of sponge-associated chlamydiae
Abstract
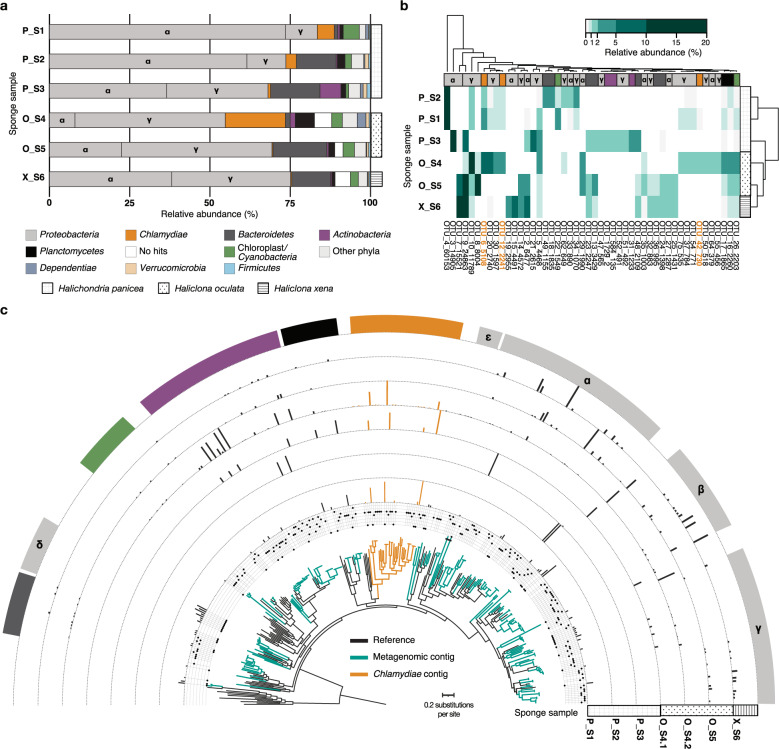
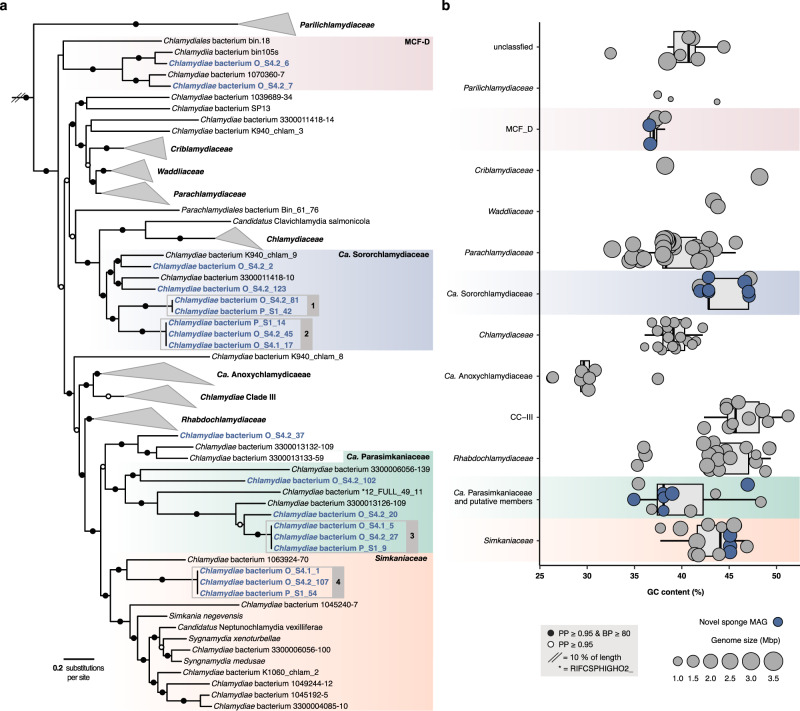
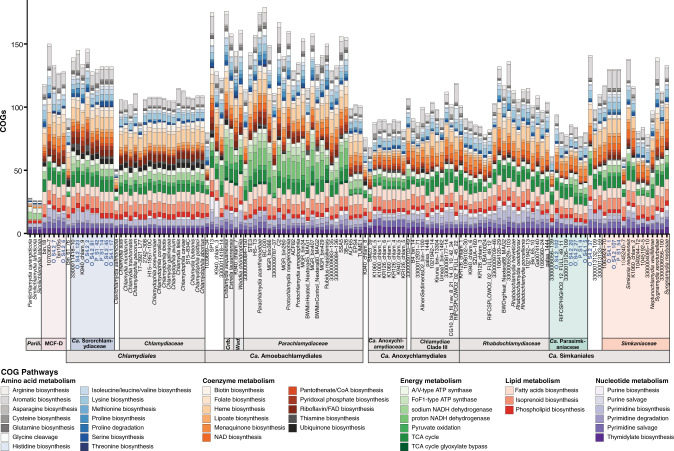
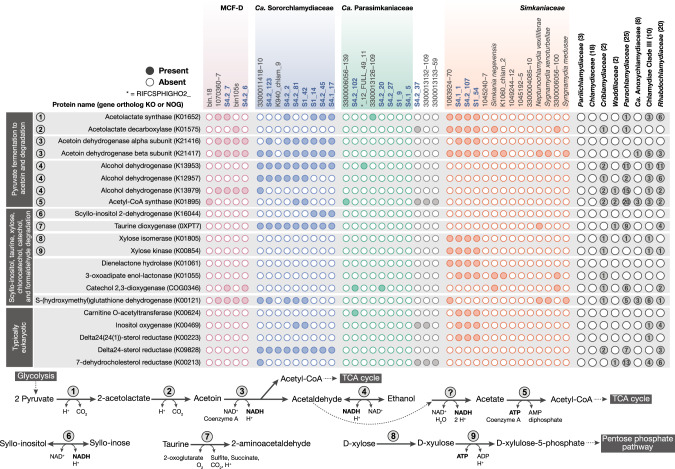
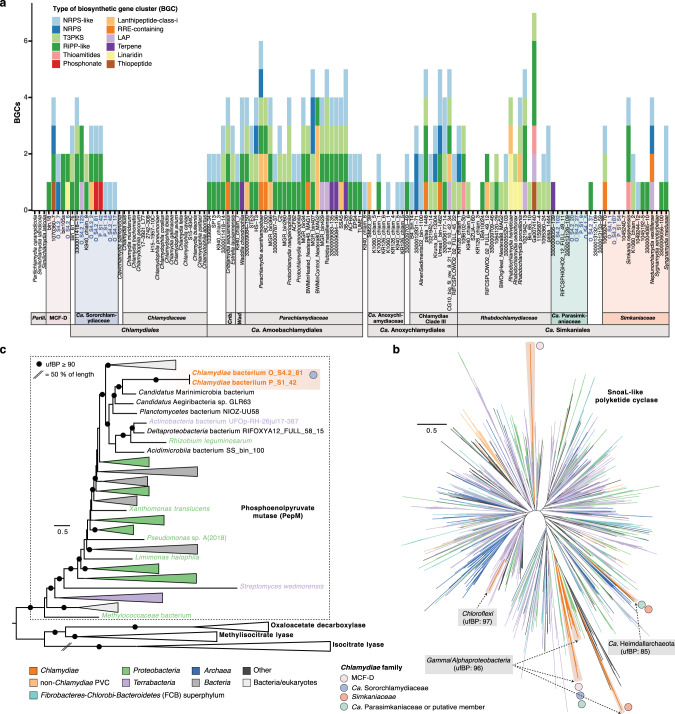
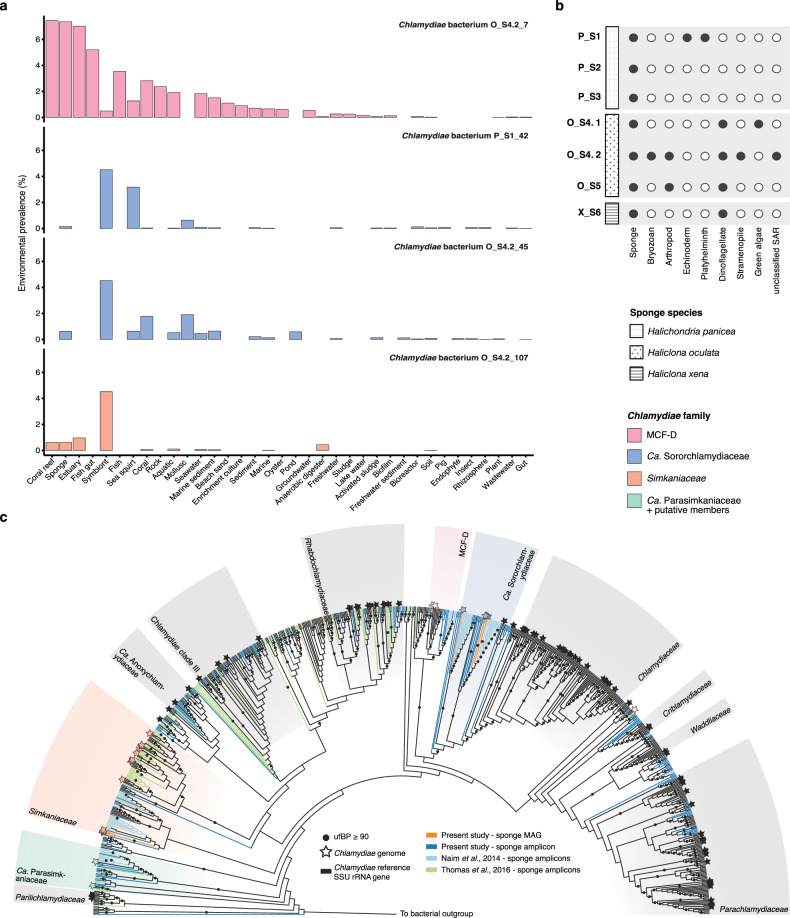
Sponge microbiomes contribute to host health, nutrition, and defense through the production of secondary metabolites. Chlamydiae, a phylum of obligate intracellular bacteria ranging from animal pathogens to endosymbionts of microbial eukaryotes, are frequently found associated with sponges. However, sponge-associated chlamydial diversity has not yet been investigated at the genomic level and host interactions thus far remain unexplored. Here, we sequenced the microbiomes of three sponge species and found high, though variable, Chlamydiae relative abundances of up to 18.7% of bacteria. Using genome-resolved metagenomics 18 high-quality sponge-associated chlamydial genomes were reconstructed, covering four chlamydial families. Among these, Candidatus Sororchlamydiaceae shares a common ancestor with Chlamydiaceae animal pathogens, suggesting long-term co-evolution with animals. Based on gene content, sponge-associated chlamydiae resemble members from the same family more than sponge-associated chlamydiae of other families, and have greater metabolic versatility than known chlamydial animal pathogens. Sponge-associated chlamydiae are also enriched in genes for degrading diverse compounds found in sponges. Unexpectedly, we identified widespread genetic potential for secondary metabolite biosynthesis across Chlamydiae, which may represent an unexplored source of novel natural products. This finding suggests that Chlamydiae members may partake in defensive symbioses and that secondary metabolites play a wider role in mediating intracellular interactions. Furthermore, sponge-associated chlamydiae relatives were found in other marine invertebrates, pointing towards wider impacts of the Chlamydiae phylum on marine ecosystems.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Marine Sediments Illuminate Chlamydiae Diversity and Evolution.Curr Biol. 2020 Mar 23;30(6):1032-1048.e7. doi: 10.1016/j.cub.2020.02.016. Epub 2020 Mar 5. Curr Biol. 2020. PMID: 32142706
-
A genomic view of trophic and metabolic diversity in clade-specific Lamellodysidea sponge microbiomes.Microbiome. 2020 Jun 23;8(1):97. doi: 10.1186/s40168-020-00877-y. Microbiome. 2020. PMID: 32576248 Free PMC article.
-
Integrating metagenomic and amplicon databases to resolve the phylogenetic and ecological diversity of the Chlamydiae.ISME J. 2014 Jan;8(1):115-25. doi: 10.1038/ismej.2013.142. Epub 2013 Aug 15. ISME J. 2014. PMID: 23949660 Free PMC article.
-
Comparative Metagenomic Analysis of Biosynthetic Diversity across Sponge Microbiomes Highlights Metabolic Novelty, Conservation, and Diversification.mSystems. 2022 Aug 30;7(4):e0035722. doi: 10.1128/msystems.00357-22. Epub 2022 Jul 18. mSystems. 2022. PMID: 35862823 Free PMC article. Review.
-
The many faces of chlamydiae.Malays J Pathol. 2000 Dec;22(2):55-64. Malays J Pathol. 2000. PMID: 16329536 Review.
Cited by
-
Trait biases in microbial reference genomes.Sci Data. 2023 Feb 9;10(1):84. doi: 10.1038/s41597-023-01994-7. Sci Data. 2023. PMID: 36759614 Free PMC article.
-
Chlamydiae in corals: shared functional potential despite broad taxonomic diversity.ISME Commun. 2024 Apr 15;4(1):ycae054. doi: 10.1093/ismeco/ycae054. eCollection 2024 Jan. ISME Commun. 2024. PMID: 38707840 Free PMC article.
-
The Fish Pathogen "Candidatus Clavichlamydia salmonicola"-A Missing Link in the Evolution of Chlamydial Pathogens of Humans.Genome Biol Evol. 2023 Aug 1;15(8):evad147. doi: 10.1093/gbe/evad147. Genome Biol Evol. 2023. PMID: 37615694 Free PMC article.
-
Colocalization and potential interactions of Endozoicomonas and chlamydiae in microbial aggregates of the coral Pocillopora acuta.Sci Adv. 2023 May 19;9(20):eadg0773. doi: 10.1126/sciadv.adg0773. Epub 2023 May 17. Sci Adv. 2023. PMID: 37196086 Free PMC article.
-
Chlamydiae as symbionts of photosynthetic dinoflagellates.ISME J. 2024 Jan 8;18(1):wrae139. doi: 10.1093/ismejo/wrae139. ISME J. 2024. PMID: 39046276 Free PMC article.
References
-
- Hentschel U, Piel J, Degnan SM, Taylor MW. Genomic insights into the marine sponge microbiome. Nat Rev Microbiol. 2012;10:641–54. - PubMed
