

The healthy equine uterus harbors a distinct core microbiome plus a rich and diverse microbiome that varies with geographical location
- PMID: 36042332
- PMCID: PMC9427864
- DOI: 10.1038/s41598-022-18971-6
The healthy equine uterus harbors a distinct core microbiome plus a rich and diverse microbiome that varies with geographical location
Abstract
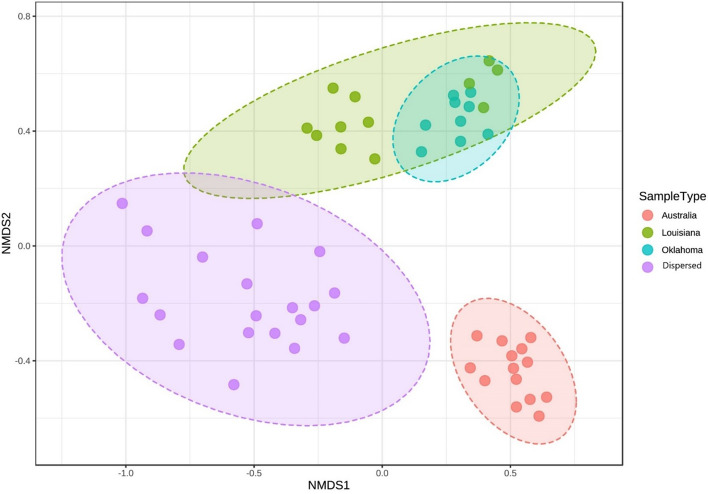
The goal of this study was to understand the composition and existence of the resident uterine microbiome in healthy mares and to establish the presence of a core microbiome for the healthy equine uterus. We analyzed the microbiomes of 35 healthy mares that are long-time residents of three farms in Oklahoma, Louisiana, and Australia as well as that of 19 mares purchased from scattered owners in the Southern Mid-Western states of the United States. Over 6 million paired-end reads of the V4 region of the 16S rRNA gene were obtained resulting in 19,542 unique Amplicon Sequence Variants (ASVs). ASVs were assigned to 17 known phyla and 213 known genera. Most abundant genera across all animals were Pseudomonas (27%) followed by Lonsdalea (8%), Lactobacillus (7.5%), Escherichia/Shigella (4.5%), and Prevotella (3%). Oklahoma and Louisiana samples were dominated by Pseudomonas (75%). Lonsdalea (28%) was the most abundant genus in the Australian samples but was not found in any other region. Microbial diversity, richness, and evenness of the equine uterine microbiome is largely dependent on the geographical location of the animal. However, we observed a core uterine microbiome consisting of Lactobacillus, Escherichia/Shigella, Streptococcus, Blautia, Staphylococcus, Klebsiella, Acinetobacter, and Peptoanaerobacter.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures







References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
