

JAG1-NOTCH4 mechanosensing drives atherosclerosis
- PMID: 36044575
- PMCID: PMC9432841
- DOI: 10.1126/sciadv.abo7958
JAG1-NOTCH4 mechanosensing drives atherosclerosis
Abstract
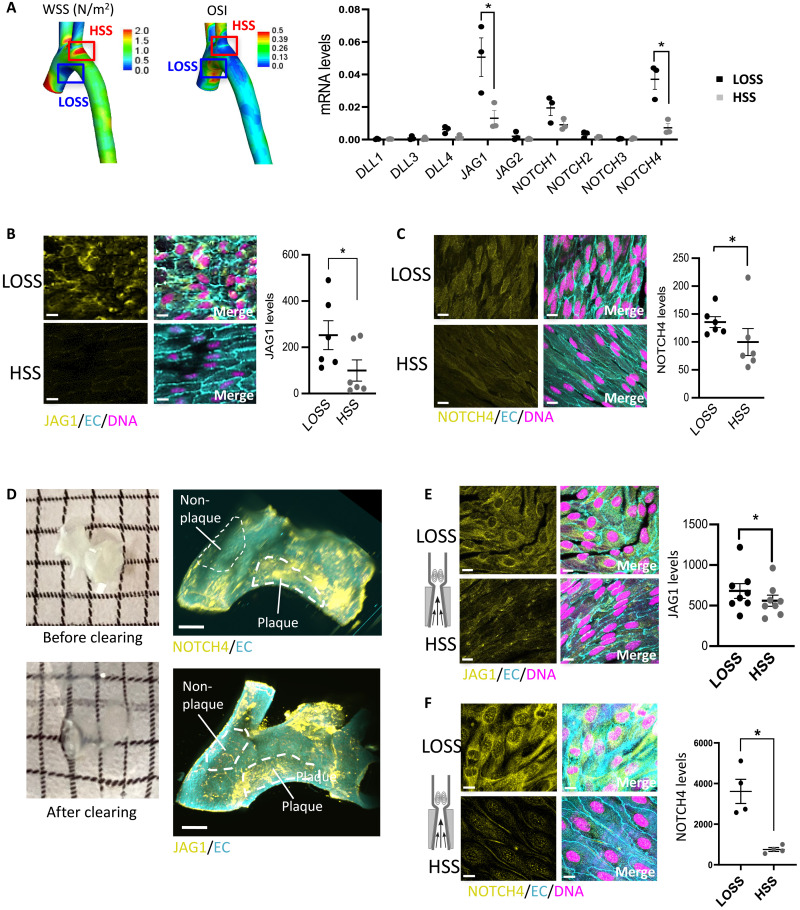
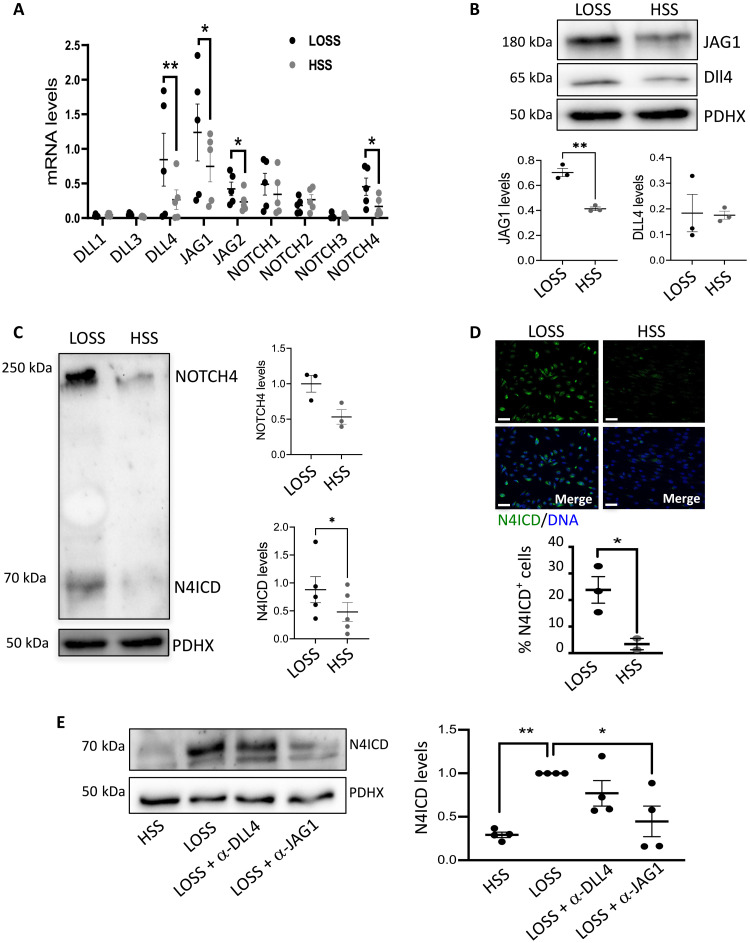
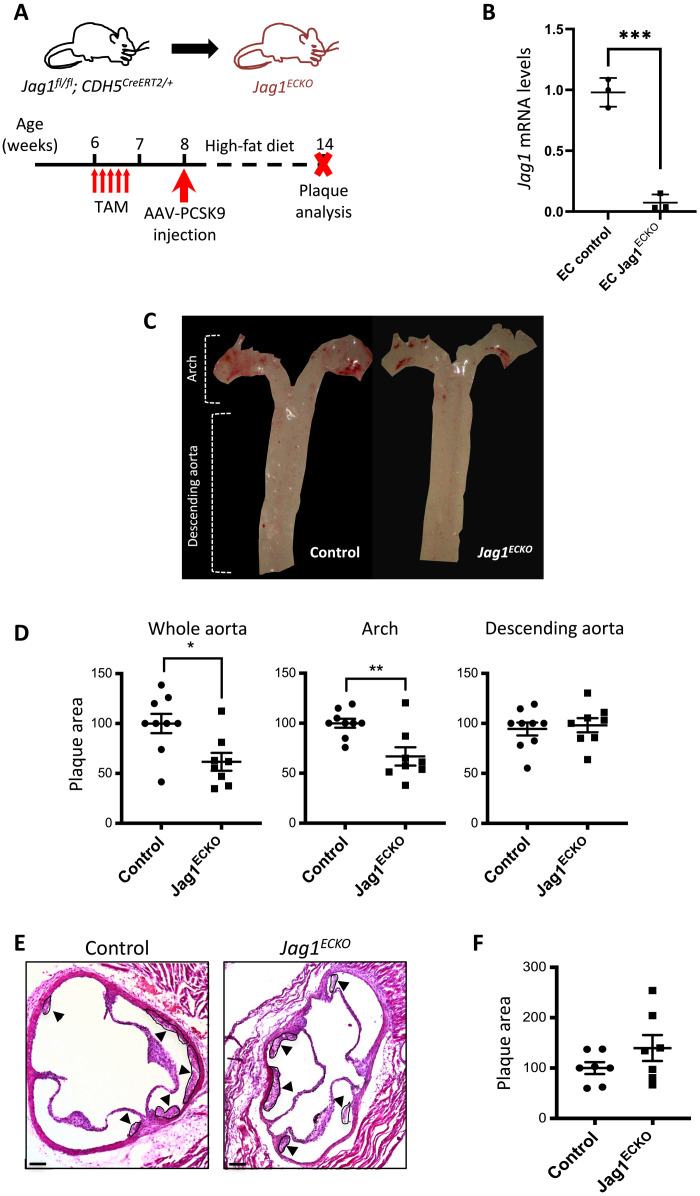
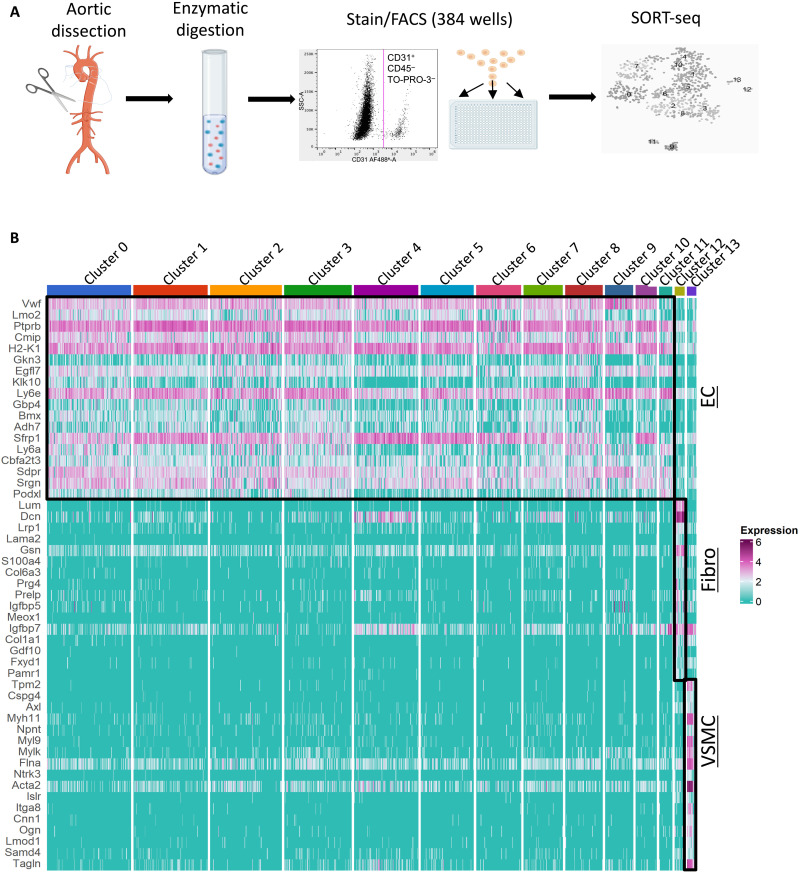
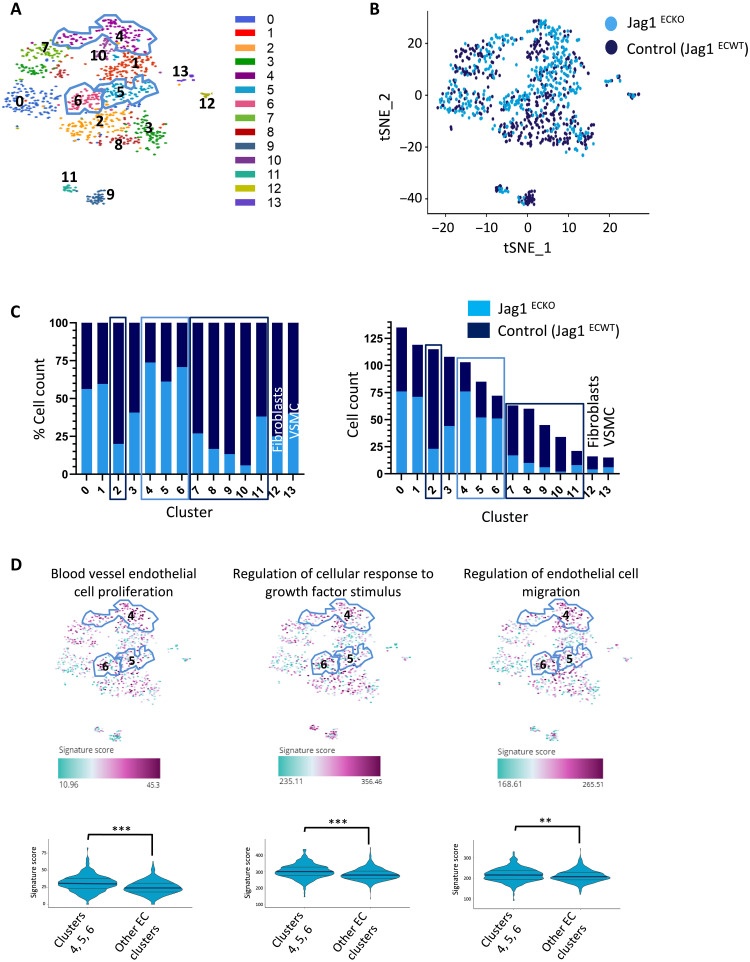
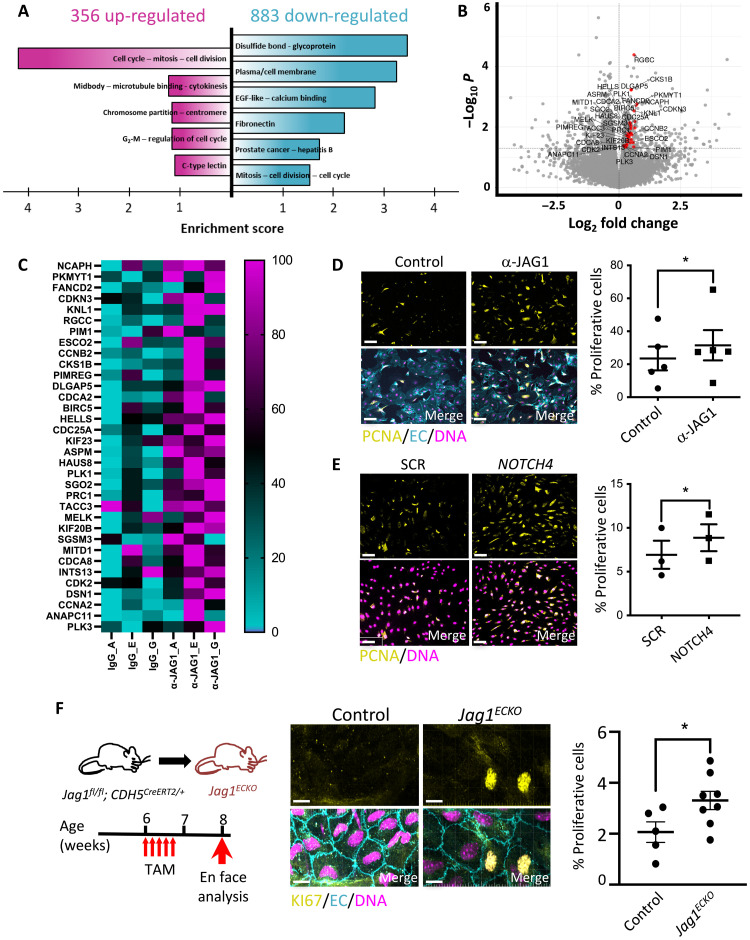
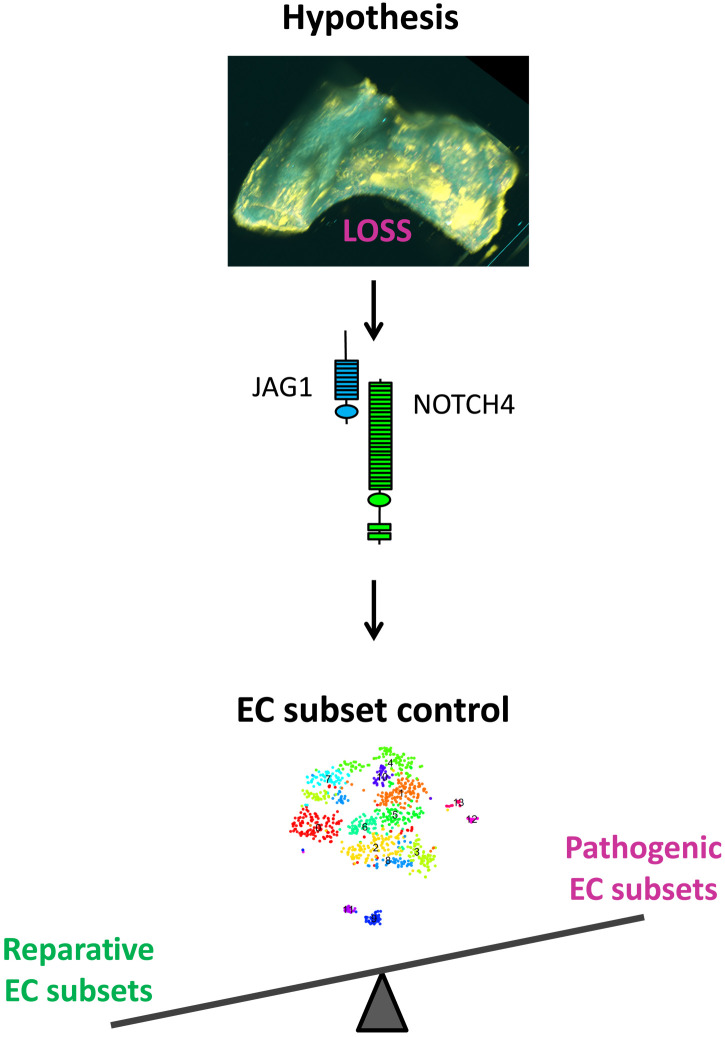
Endothelial cell (EC) sensing of disturbed blood flow triggers atherosclerosis, a disease of arteries that causes heart attack and stroke, through poorly defined mechanisms. The Notch pathway plays a central role in blood vessel growth and homeostasis, but its potential role in sensing of disturbed flow has not been previously studied. Here, we show using porcine and murine arteries and cultured human coronary artery EC that disturbed flow activates the JAG1-NOTCH4 signaling pathway. Light-sheet imaging revealed enrichment of JAG1 and NOTCH4 in EC of atherosclerotic plaques, and EC-specific genetic deletion of Jag1 (Jag1ECKO) demonstrated that Jag1 promotes atherosclerosis at sites of disturbed flow. Mechanistically, single-cell RNA sequencing in Jag1ECKO mice demonstrated that Jag1 suppresses subsets of ECs that proliferate and migrate. We conclude that JAG1-NOTCH4 sensing of disturbed flow enhances atherosclerosis susceptibility by regulating EC heterogeneity and that therapeutic targeting of this pathway may treat atherosclerosis.
Figures
References
-
- Souilhol C., Serbanovic-Canic J., Fragiadaki M., Chico T. J., Ridger V., Roddie H., Evans P. C., Endothelial responses to shear stress in atherosclerosis: A novel role for developmental genes. Nat. Rev. Cardiol. 17, 52–63 (2019). - PubMed
-
- Brown A. J., Teng Z. Z., Evans P. C., Gillard J. H., Samady H., Bennett M. R., Role of biomechanical forces in the natural history of coronary atherosclerosis. Nat. Rev. Cardiol. 13, 210–220 (2016). - PubMed
-
- Kwak B. R., Bäck M., Bochaton-Piallat M. L., Caligiuri G., Daemens M., Davies P. F., Hoefer I. E., Holvoet P., Jo H., Krams R., Lehoux S., Monaco C., Steffens S., Virmani R., Weber C., Wentzel J. J., Evans P. C., Biomechanical factors in atherosclerosis: Mechanisms and clinical implications. Eur. Heart J. 35, 3013–3020, 3020a-3020d (2014). - PMC - PubMed
-
- Artavanis-Tsakonas S., Rand M. D., Lake R. J., Notch signaling: Cell fate control and signal integration in development. Science 284, 770–776 (1999). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
