

Bi-allelic loss-of-function variants in TMEM147 cause moderate to profound intellectual disability with facial dysmorphism and pseudo-Pelger-Huët anomaly
- PMID: 36044892
- PMCID: PMC9606387
- DOI: 10.1016/j.ajhg.2022.08.008
Bi-allelic loss-of-function variants in TMEM147 cause moderate to profound intellectual disability with facial dysmorphism and pseudo-Pelger-Huët anomaly
Abstract
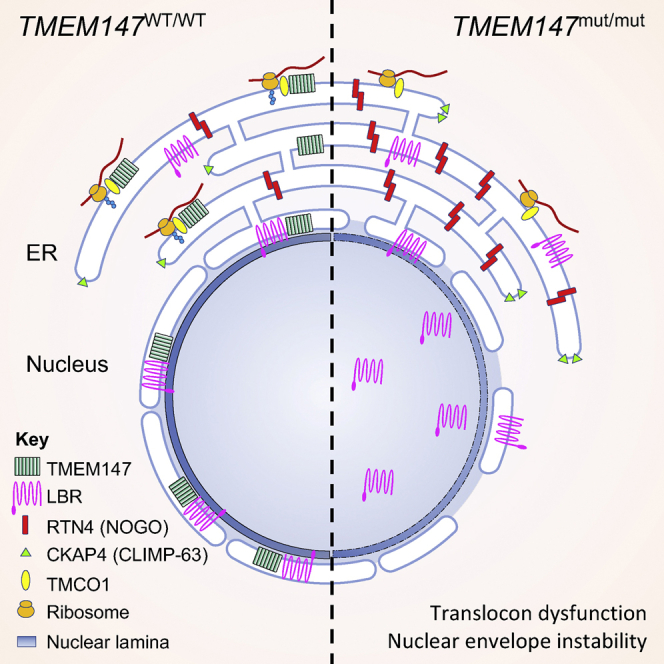
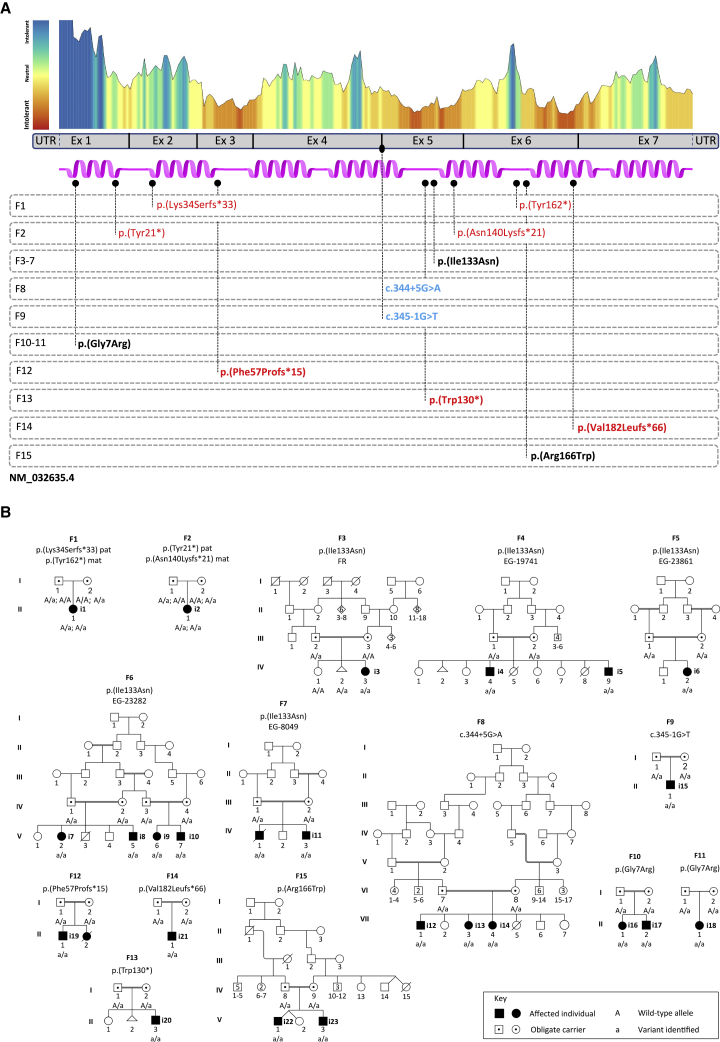
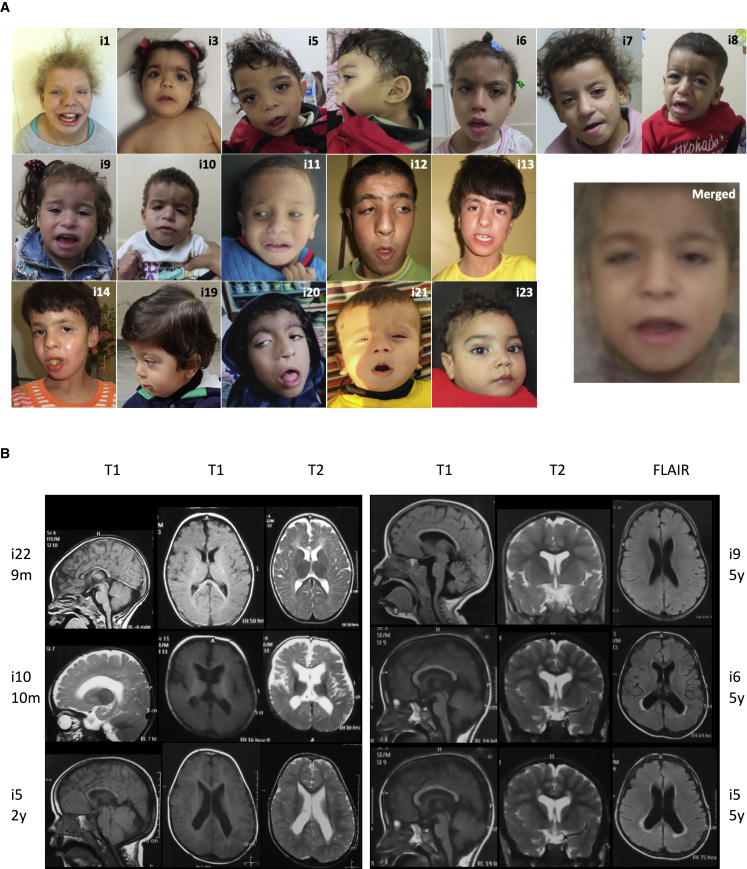
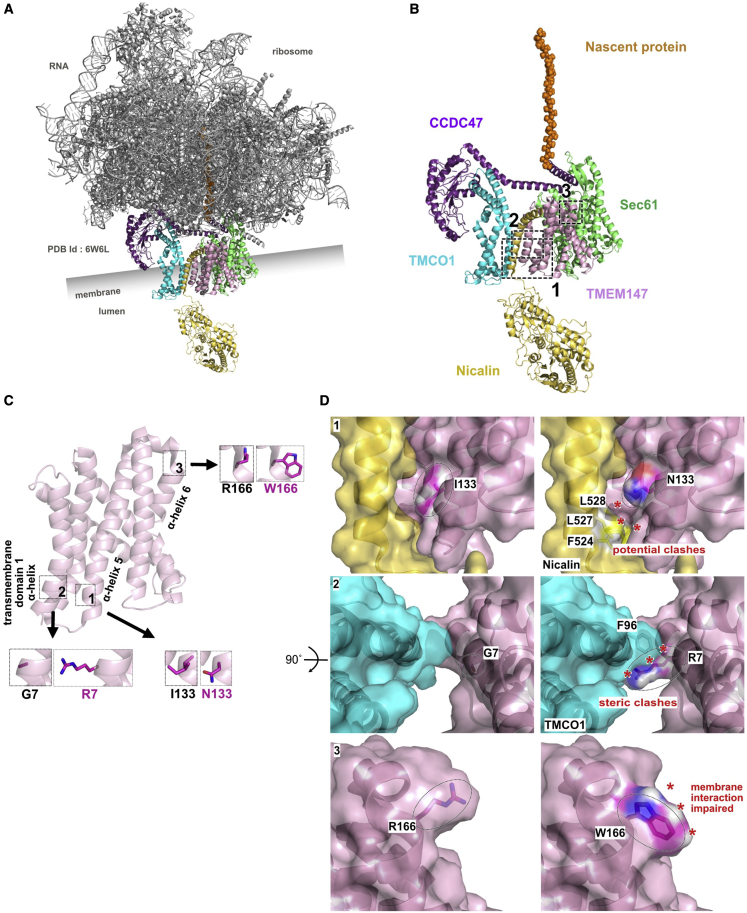
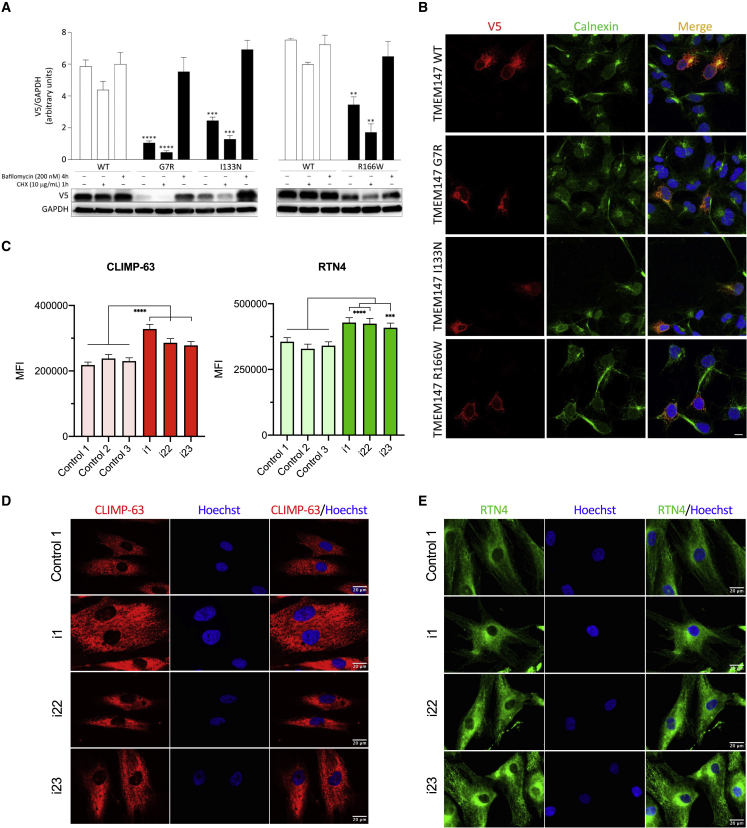
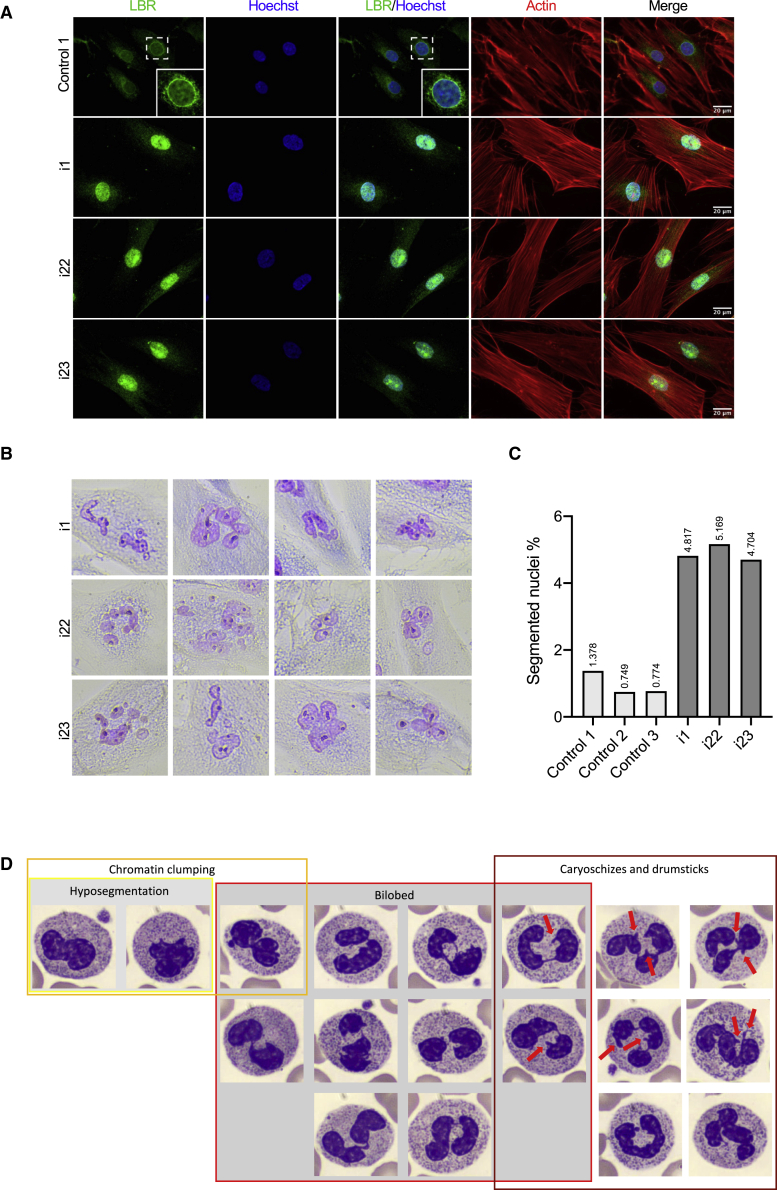
The transmembrane protein TMEM147 has a dual function: first at the nuclear envelope, where it anchors lamin B receptor (LBR) to the inner membrane, and second at the endoplasmic reticulum (ER), where it facilitates the translation of nascent polypeptides within the ribosome-bound TMCO1 translocon complex. Through international data sharing, we identified 23 individuals from 15 unrelated families with bi-allelic TMEM147 loss-of-function variants, including splice-site, nonsense, frameshift, and missense variants. These affected children displayed congruent clinical features including coarse facies, developmental delay, intellectual disability, and behavioral problems. In silico structural analyses predicted disruptive consequences of the identified amino acid substitutions on translocon complex assembly and/or function, and in vitro analyses documented accelerated protein degradation via the autophagy-lysosomal-mediated pathway. Furthermore, TMEM147-deficient cells showed CKAP4 (CLIMP-63) and RTN4 (NOGO) upregulation with a concomitant reorientation of the ER, which was also witnessed in primary fibroblast cell culture. LBR mislocalization and nuclear segmentation was observed in primary fibroblast cells. Abnormal nuclear segmentation and chromatin compaction were also observed in approximately 20% of neutrophils, indicating the presence of a pseudo-Pelger-Huët anomaly. Finally, co-expression analysis revealed significant correlation with neurodevelopmental genes in the brain, further supporting a role of TMEM147 in neurodevelopment. Our findings provide clinical, genetic, and functional evidence that bi-allelic loss-of-function variants in TMEM147 cause syndromic intellectual disability due to ER-translocon and nuclear organization dysfunction.
Keywords: DNA methylation; LBR; Pelger-Huët anomaly; TMEM147; facial dysmorphism; intellectual disability; neurodevelopmental disorder; nuclear envelope instability; transcriptomics; translocon dysfunction.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests S.A., I.K., J.P.B., and A.B.-A. are employees of Centogene GmbH.
Figures
Similar articles
-
A biallelic loss-of-function variant in TMEM147 causes profound intellectual disability and spasticity.Neurogenetics. 2023 Oct;24(4):311-316. doi: 10.1007/s10048-023-00734-8. Epub 2023 Sep 5. Neurogenetics. 2023. PMID: 37668766
-
[Nuclear abnormalities in Pelger-Huet anomaly; progress in blood cell morphology].Rinsho Byori. 2005 Jan;53(1):54-60. Rinsho Byori. 2005. PMID: 15724491 Review. Japanese.
-
Lamin B-receptor mutations in Pelger-Huët anomaly.Br J Haematol. 2003 Nov;123(3):542-4. doi: 10.1046/j.1365-2141.2003.04621.x. Br J Haematol. 2003. PMID: 14617022
-
Mutations causing Greenberg dysplasia but not Pelger anomaly uncouple enzymatic from structural functions of a nuclear membrane protein.Nucleus. 2010 Jul-Aug;1(4):354-66. doi: 10.4161/nucl.1.4.12435. Epub 2010 May 21. Nucleus. 2010. PMID: 21327084 Free PMC article.
-
Craniofacial dysmorphism, skeletal anomalies, and impaired intellectual development syndrome-1 in two new patients with the same homozygous TMCO1 variant and review of the literature.Eur J Med Genet. 2023 Mar;66(3):104715. doi: 10.1016/j.ejmg.2023.104715. Epub 2023 Jan 25. Eur J Med Genet. 2023. PMID: 36708876 Review.
Cited by
-
Panoramic variation analysis of a family with neurodevelopmental disorders caused by biallelic loss-of-function variants in TMEM141, DDHD2, and LHFPL5.Front Med. 2024 Feb;18(1):81-97. doi: 10.1007/s11684-023-1006-x. Epub 2023 Oct 14. Front Med. 2024. PMID: 37837560
-
OMIXCARE: OMICS technologies solved about 33% of the patients with heterogeneous rare neuro-developmental disorders and negative exome sequencing results and identified 13% additional candidate variants.Front Cell Dev Biol. 2022 Oct 28;10:1021785. doi: 10.3389/fcell.2022.1021785. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36393831 Free PMC article.
-
A biallelic loss-of-function variant in TMEM147 causes profound intellectual disability and spasticity.Neurogenetics. 2023 Oct;24(4):311-316. doi: 10.1007/s10048-023-00734-8. Epub 2023 Sep 5. Neurogenetics. 2023. PMID: 37668766
-
An update on autophagy disorders.J Inherit Metab Dis. 2025 Jan;48(1):e12798. doi: 10.1002/jimd.12798. Epub 2024 Oct 17. J Inherit Metab Dis. 2025. PMID: 39420677 Free PMC article. Review.
-
Loss of the endoplasmic reticulum protein Tmem208 affects cell polarity, development, and viability.Proc Natl Acad Sci U S A. 2024 Feb 27;121(9):e2322582121. doi: 10.1073/pnas.2322582121. Epub 2024 Feb 21. Proc Natl Acad Sci U S A. 2024. PMID: 38381787 Free PMC article.
References
-
- Schalock R.L., Luckasson R., Tassé M.J. An Overview of intellectual disability: Definition, diagnosis, classification, and systems of supports (12th ed.) Am. J. Intellect. Dev. Disabil. 2021;126:439–442. - PubMed
-
- Alazami A.M., Patel N., Shamseldin H.E., Anazi S., Al-Dosari M.S., Alzahrani F., Hijazi H., Alshammari M., Aldahmesh M.A., Salih M.A., et al. Accelerating Novel Candidate Gene Discovery in Neurogenetic Disorders via Whole-Exome Sequencing of Prescreened Multiplex Consanguineous Families. Cell Rep. 2015;10:148–161. - PubMed
-
- Anazi S., Maddirevula S., Salpietro V., Asi Y.T., Alsahli S., Alhashem A., Shamseldin H.E., AlZahrani F., Patel N., Ibrahim N., et al. Expanding the genetic heterogeneity of intellectual disability. Hum. Genet. 2017;136:1419–1429. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
