

African-specific molecular taxonomy of prostate cancer
- PMID: 36045292
- PMCID: PMC9477733
- DOI: 10.1038/s41586-022-05154-6
African-specific molecular taxonomy of prostate cancer
Abstract
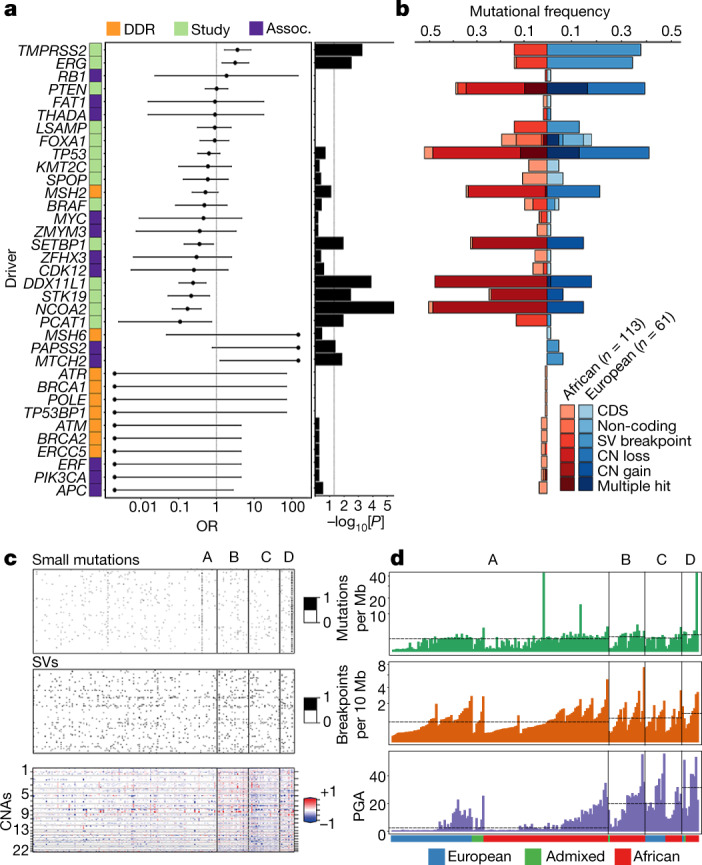
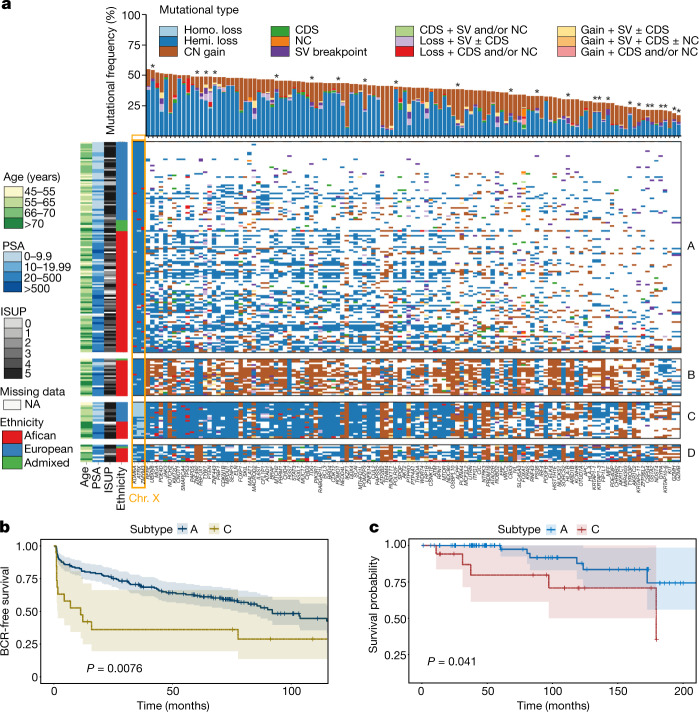
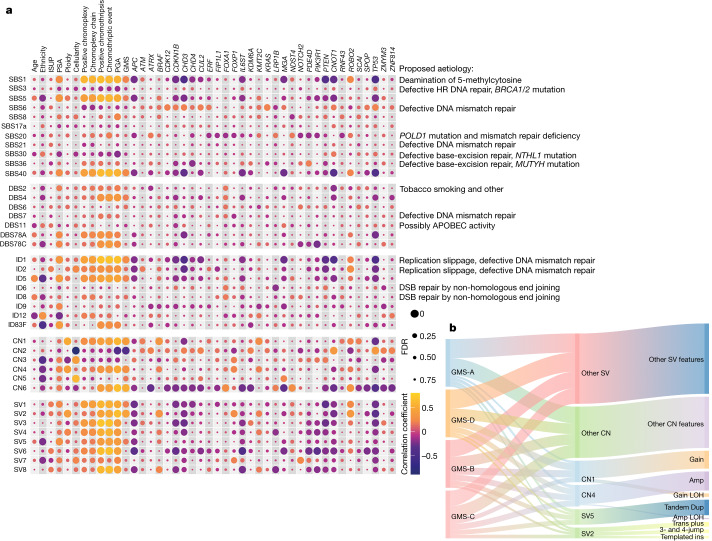
Prostate cancer is characterized by considerable geo-ethnic disparity. African ancestry is a significant risk factor, with mortality rates across sub-Saharan Africa of 2.7-fold higher than global averages1. The contributing genetic and non-genetic factors, and associated mutational processes, are unknown2,3. Here, through whole-genome sequencing of treatment-naive prostate cancer samples from 183 ancestrally (African versus European) and globally distinct patients, we generate a large cancer genomics resource for sub-Saharan Africa, identifying around 2 million somatic variants. Significant African-ancestry-specific findings include an elevated tumour mutational burden, increased percentage of genome alteration, a greater number of predicted damaging mutations and a higher total of mutational signatures, and the driver genes NCOA2, STK19, DDX11L1, PCAT1 and SETBP1. Examining all somatic mutational types, we describe a molecular taxonomy for prostate cancer differentiated by ancestry and defined as global mutational subtypes (GMS). By further including Chinese Asian data, we confirm that GMS-B (copy-number gain) and GMS-D (mutationally noisy) are specific to African populations, GMS-A (mutationally quiet) is universal (all ethnicities) and the African-European-restricted subtype GMS-C (copy-number losses) predicts poor clinical outcomes. In addition to the clinical benefit of including individuals of African ancestry, our GMS subtypes reveal different evolutionary trajectories and mutational processes suggesting that both common genetic and environmental factors contribute to the disparity between ethnicities. Analogous to gene-environment interaction-defined here as a different effect of an environmental surrounding in people with different ancestries or vice versa-we anticipate that GMS subtypes act as a proxy for intrinsic and extrinsic mutational processes in cancers, promoting global inclusion in landmark studies.
© 2022. Crown.
Conflict of interest statement
The authors declare no competing interests.
Figures












Comment in
-
Ancestry-defined molecular taxonomy of prostate cancer.Trends Cancer. 2022 Dec;8(12):973-975. doi: 10.1016/j.trecan.2022.10.008. Epub 2022 Nov 3. Trends Cancer. 2022. PMID: 36335083
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
