

Current strategies for the design of PROTAC linkers: a critical review
- PMID: 36046485
- PMCID: PMC9400730
- DOI: 10.37349/etat.2020.00018
Current strategies for the design of PROTAC linkers: a critical review
Abstract
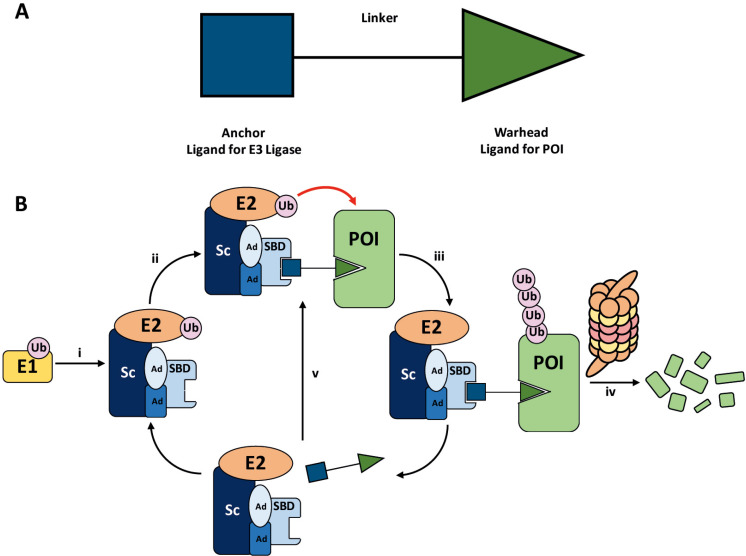
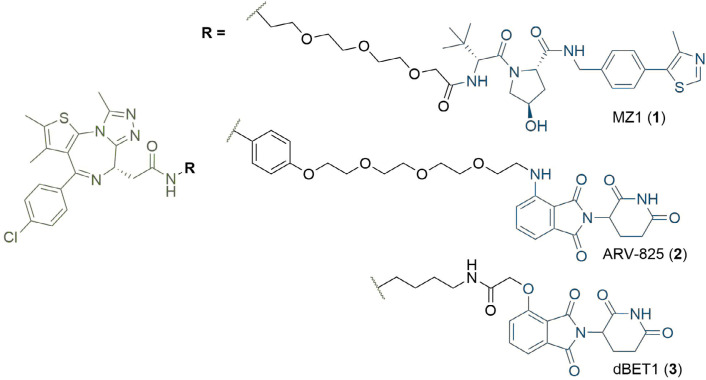
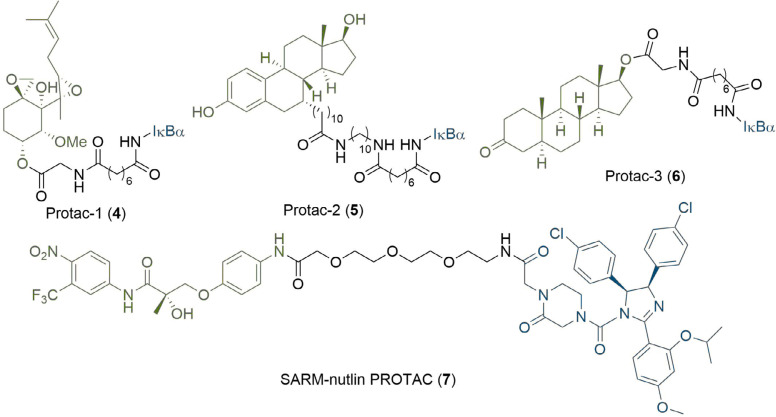
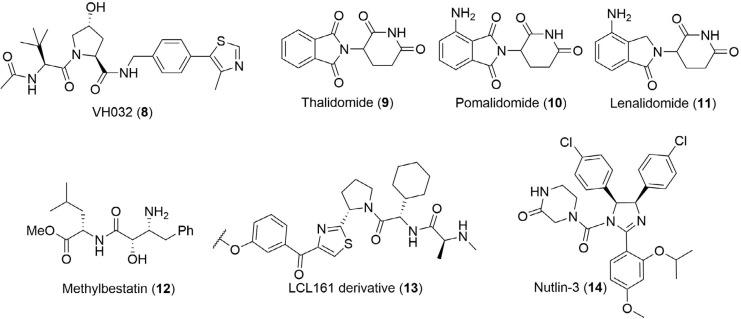
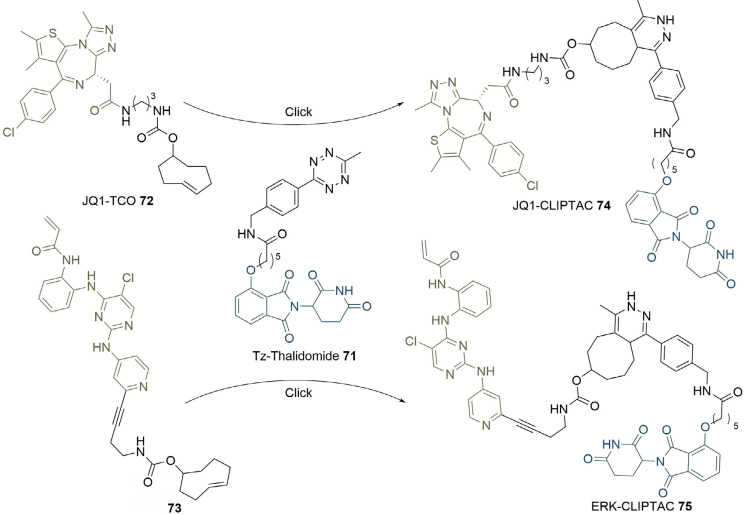
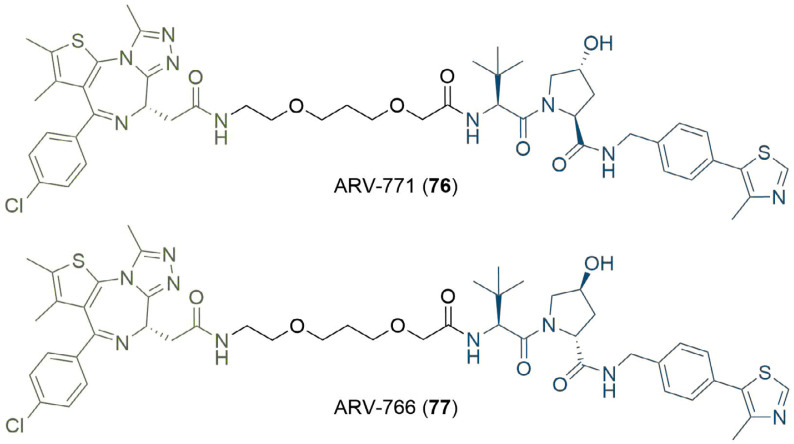
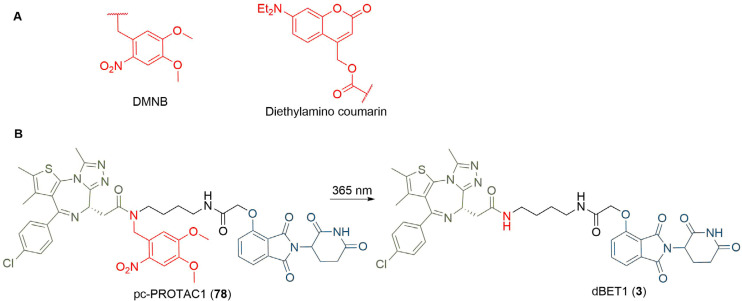
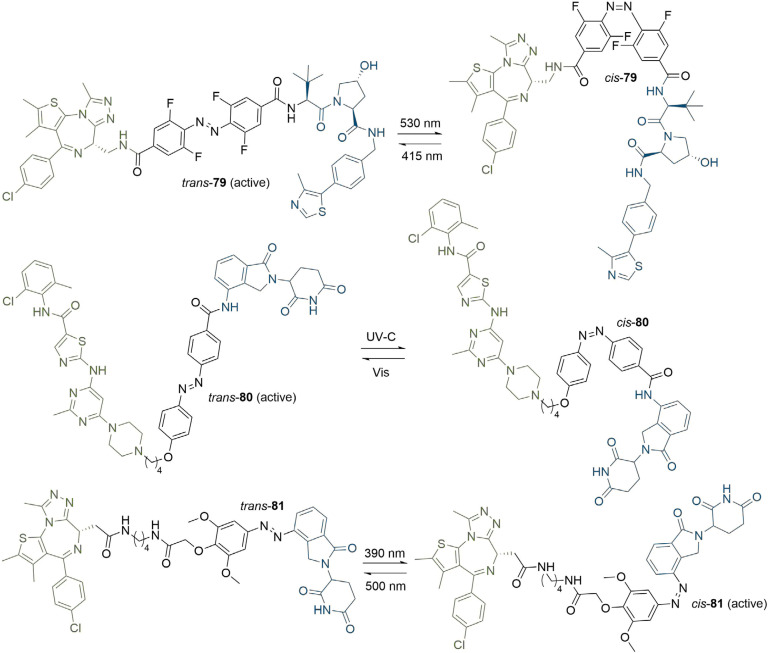
PROteolysis TArgeting Chimeras (PROTACs) are heterobifunctional molecules consisting of two ligands; an "anchor" to bind to an E3 ubiquitin ligase and a "warhead" to bind to a protein of interest, connected by a chemical linker. Targeted protein degradation by PROTACs has emerged as a new modality for the knock down of a range of proteins, with the first agents now reaching clinical evaluation. It has become increasingly clear that the length and composition of the linker play critical roles on the physicochemical properties and bioactivity of PROTACs. While linker design has historically received limited attention, the PROTAC field is evolving rapidly and currently undergoing an important shift from synthetically tractable alkyl and polyethylene glycol to more sophisticated functional linkers. This promises to unlock a wealth of novel PROTAC agents with enhanced bioactivity for therapeutic intervention. Here, the authors provide a timely overview of the diverse linker classes in the published literature, along with their underlying design principles and overall influence on the properties and bioactivity of the associated PROTACs. Finally, the authors provide a critical analysis of current strategies for PROTAC assembly. The authors highlight important limitations associated with the traditional "trial and error" approach around linker design and selection, and suggest potential future avenues to further inform rational linker design and accelerate the identification of optimised PROTACs. In particular, the authors believe that advances in computational and structural methods will play an essential role to gain a better understanding of the structure and dynamics of PROTAC ternary complexes, and will be essential to address the current gaps in knowledge associated with PROTAC design.
Keywords: PROTAC; linker design; protein degradation.
© The Author(s) 2020.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
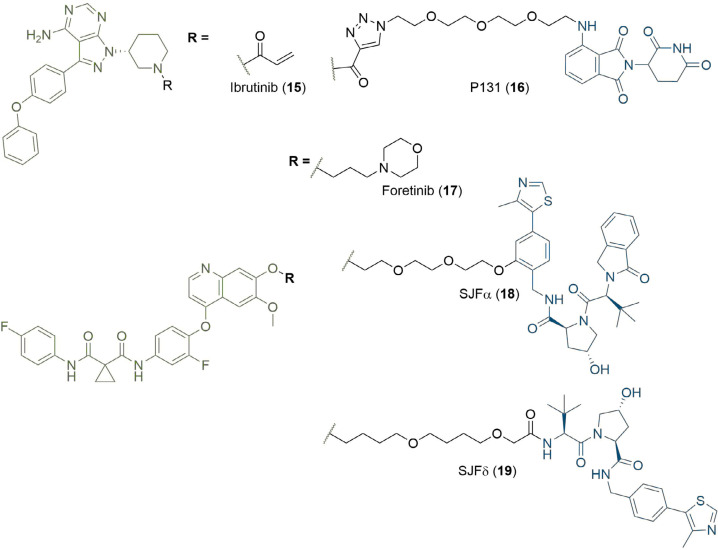
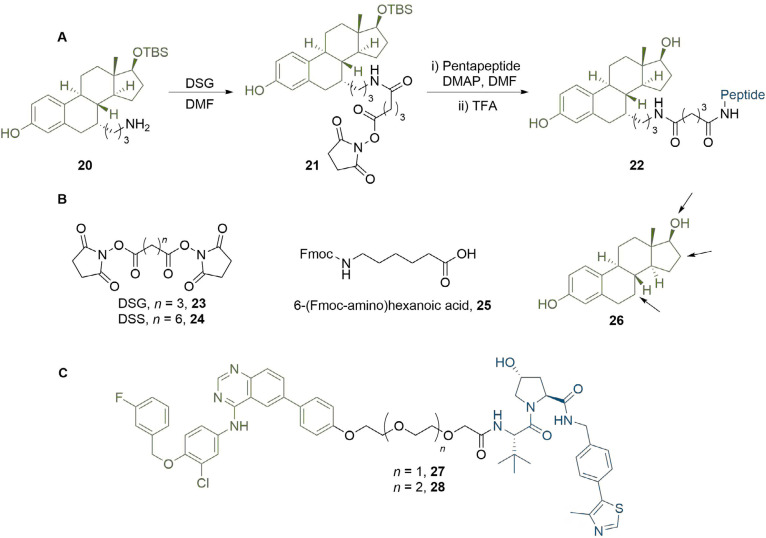
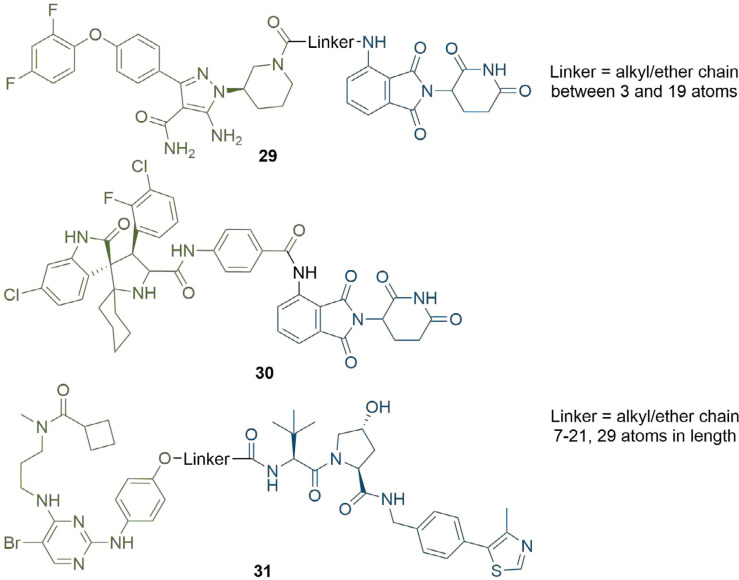
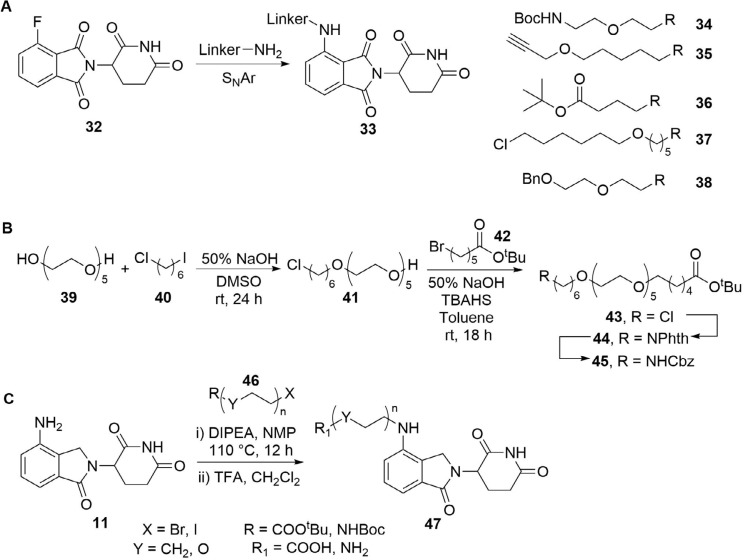
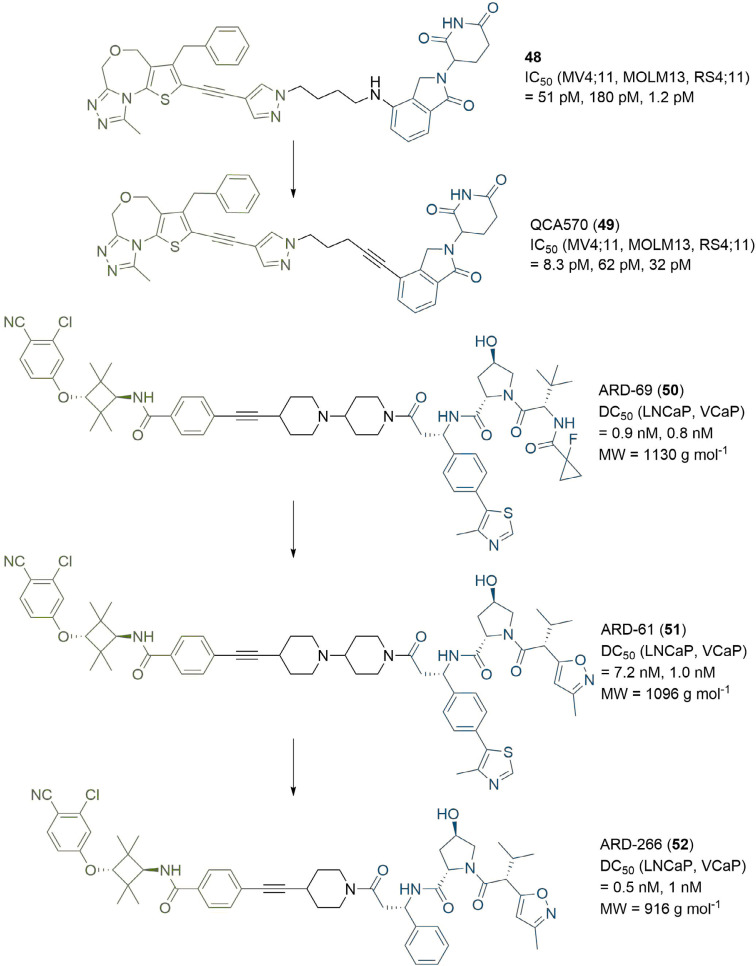
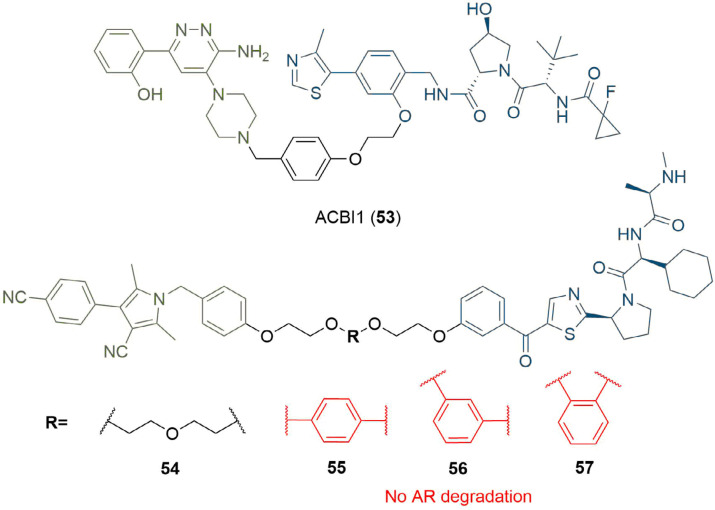
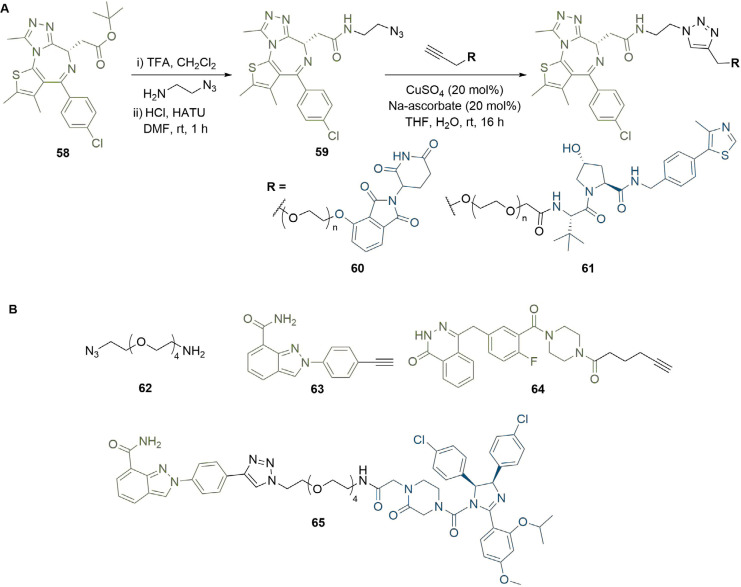
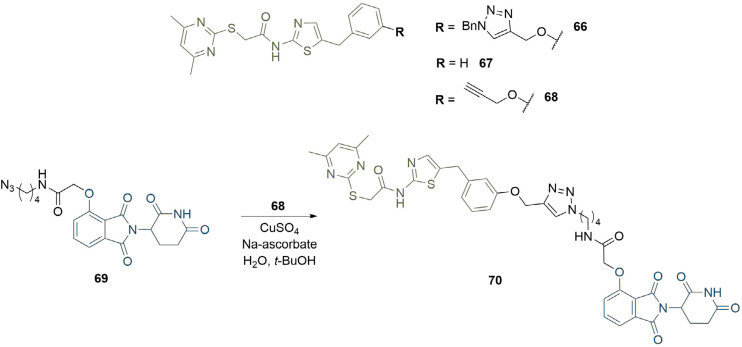
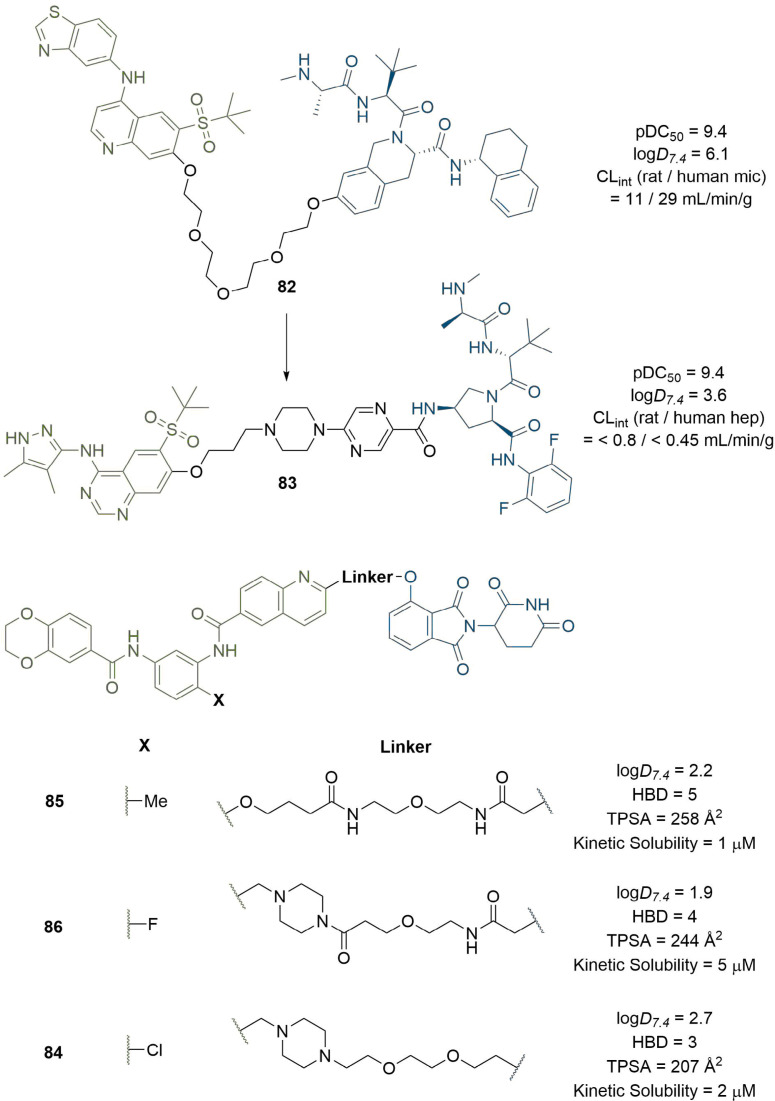
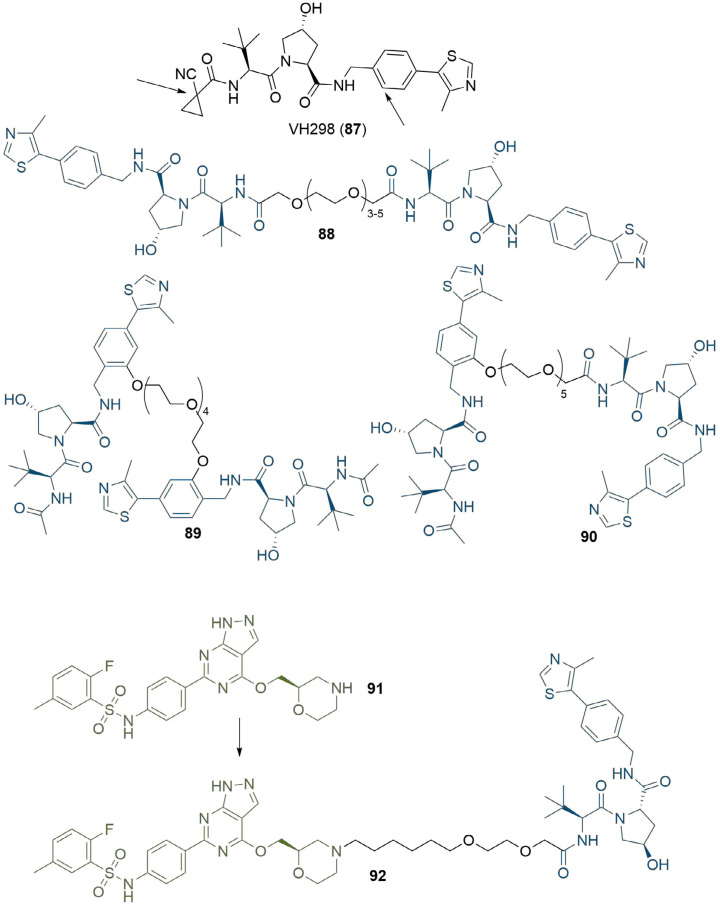
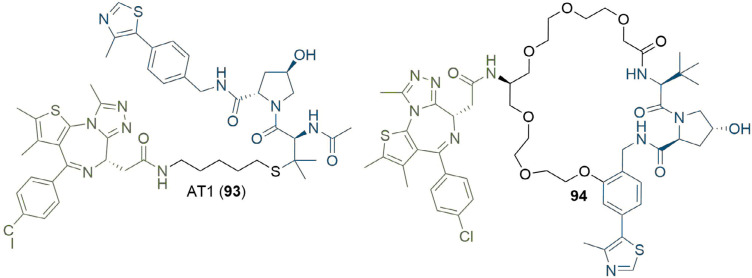
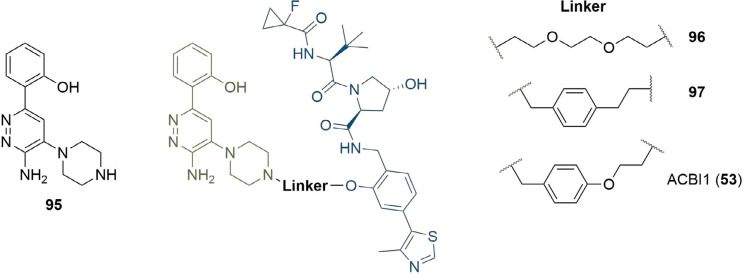
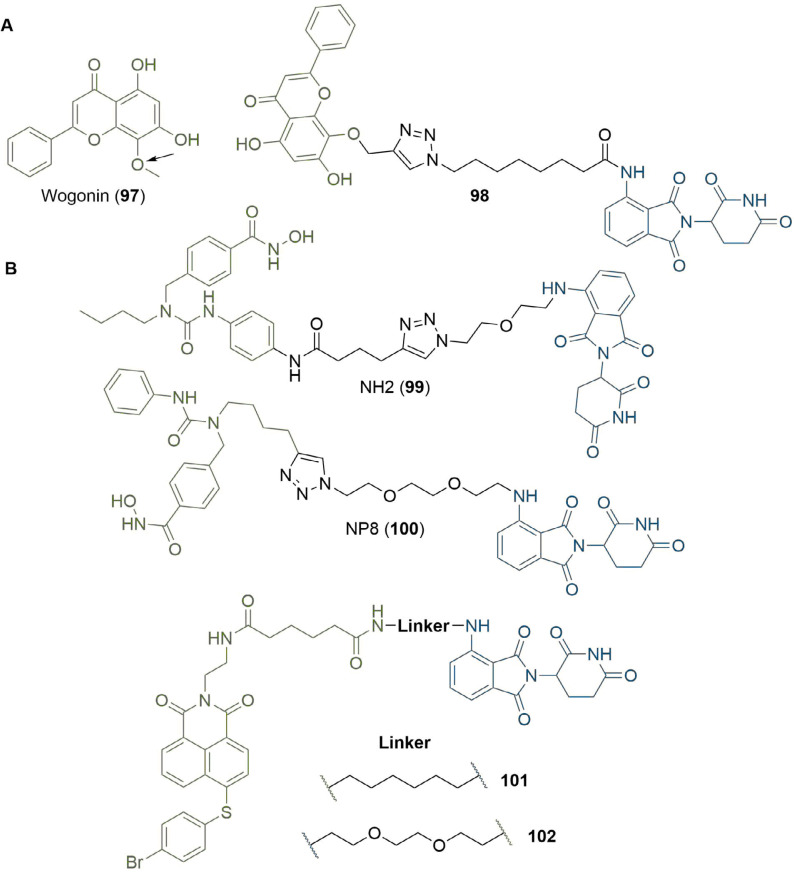
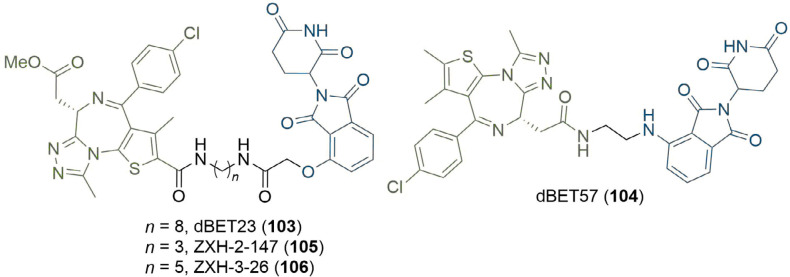
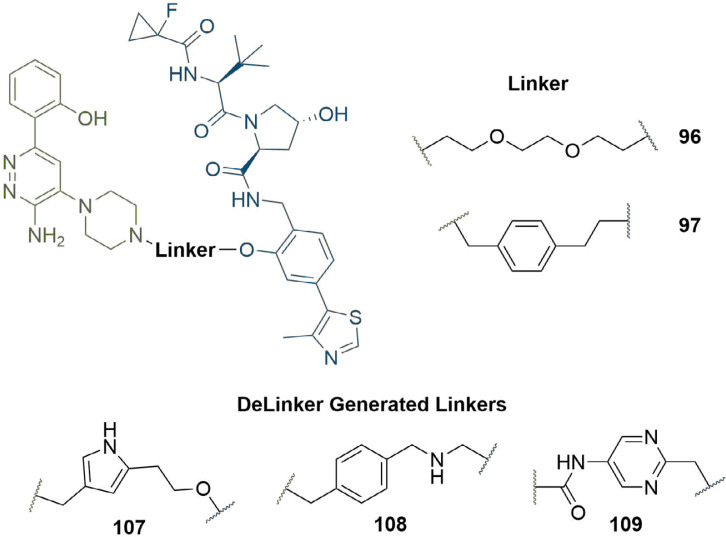
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources