

Hypoxia-Driven M2-Polarized Macrophages Facilitate Cancer Aggressiveness and Temozolomide Resistance in Glioblastoma
- PMID: 36046687
- PMCID: PMC9423979
- DOI: 10.1155/2022/1614336
Hypoxia-Driven M2-Polarized Macrophages Facilitate Cancer Aggressiveness and Temozolomide Resistance in Glioblastoma
Abstract
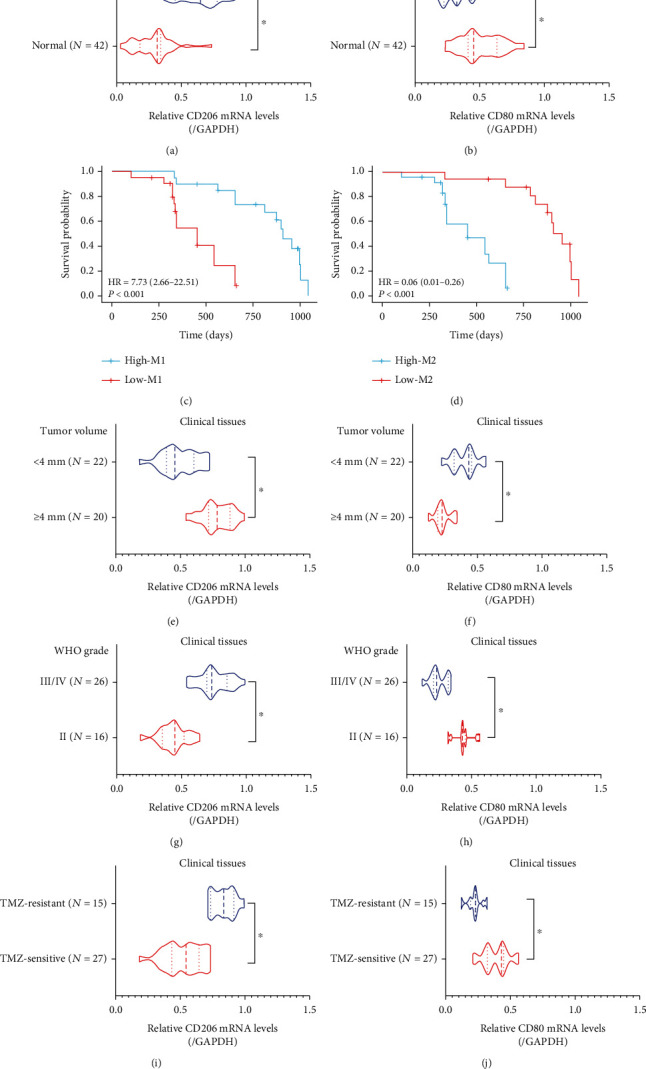
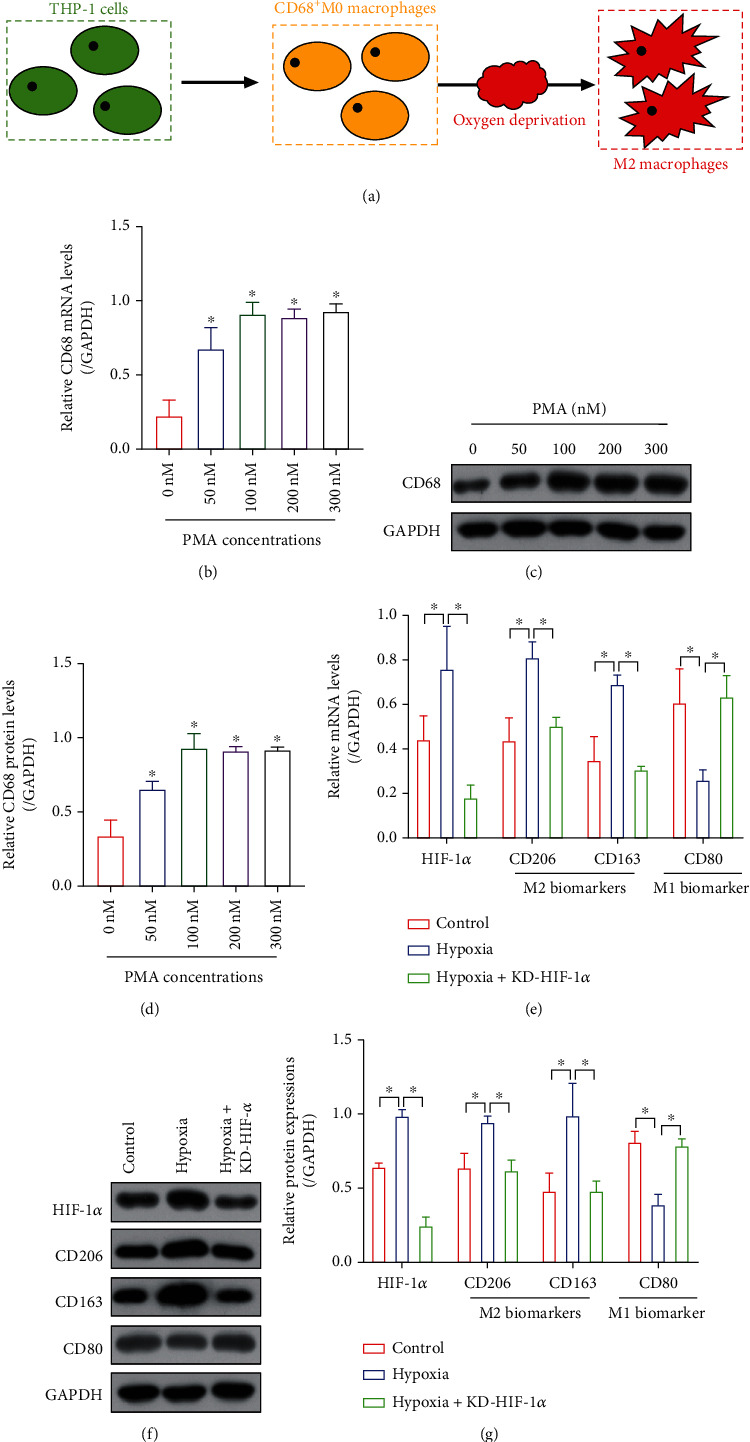
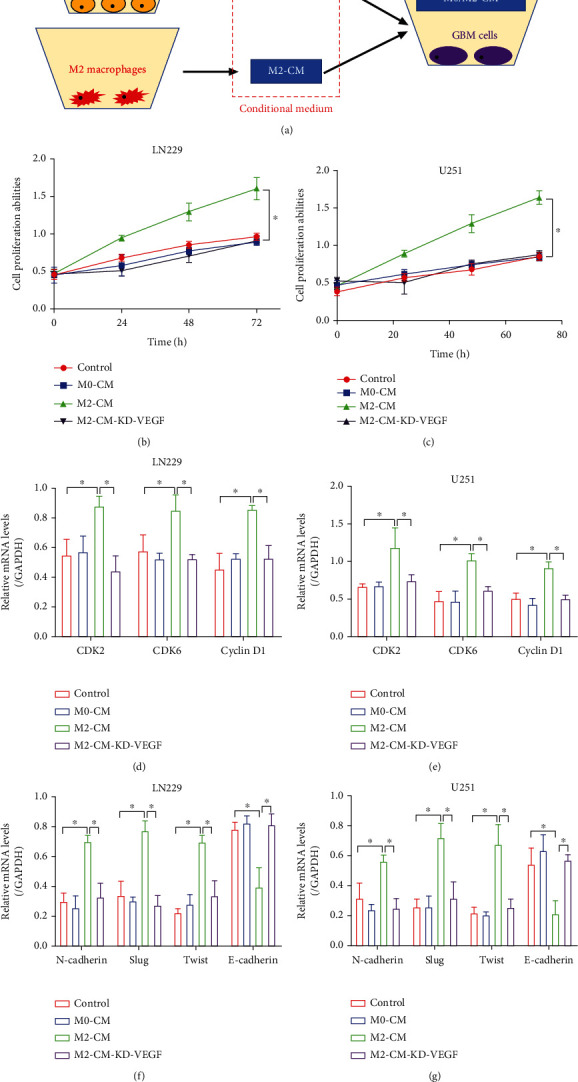
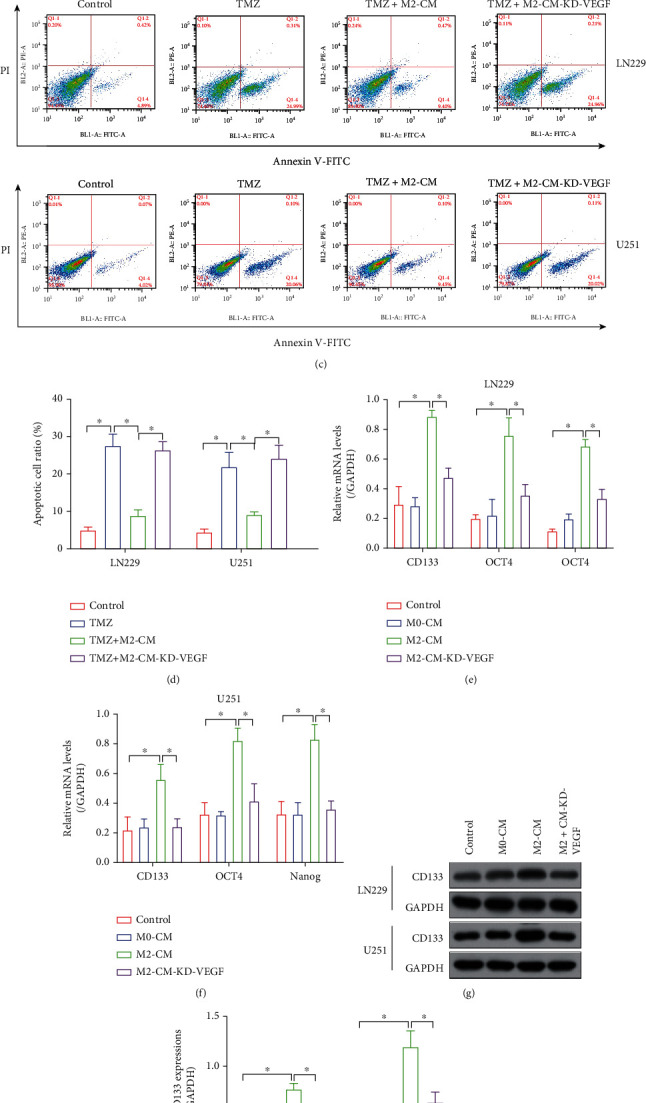
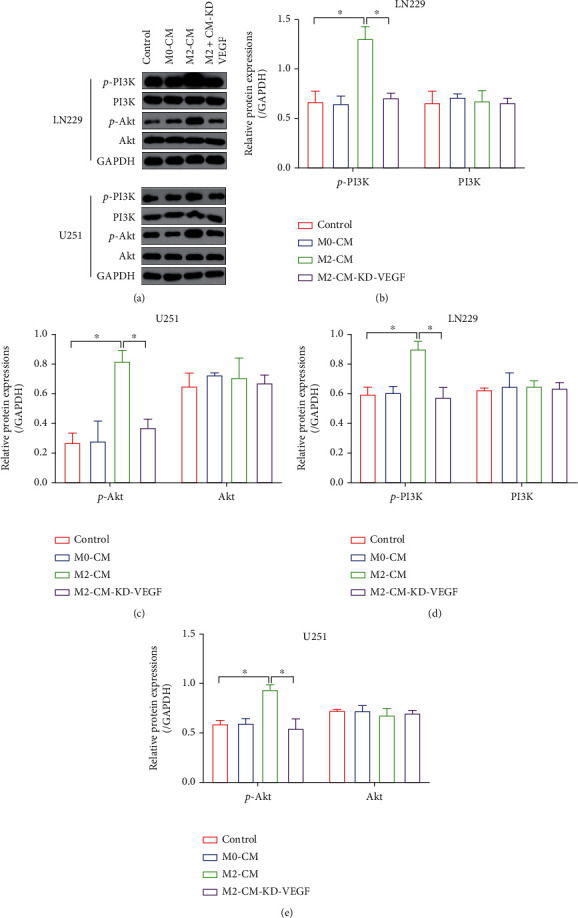
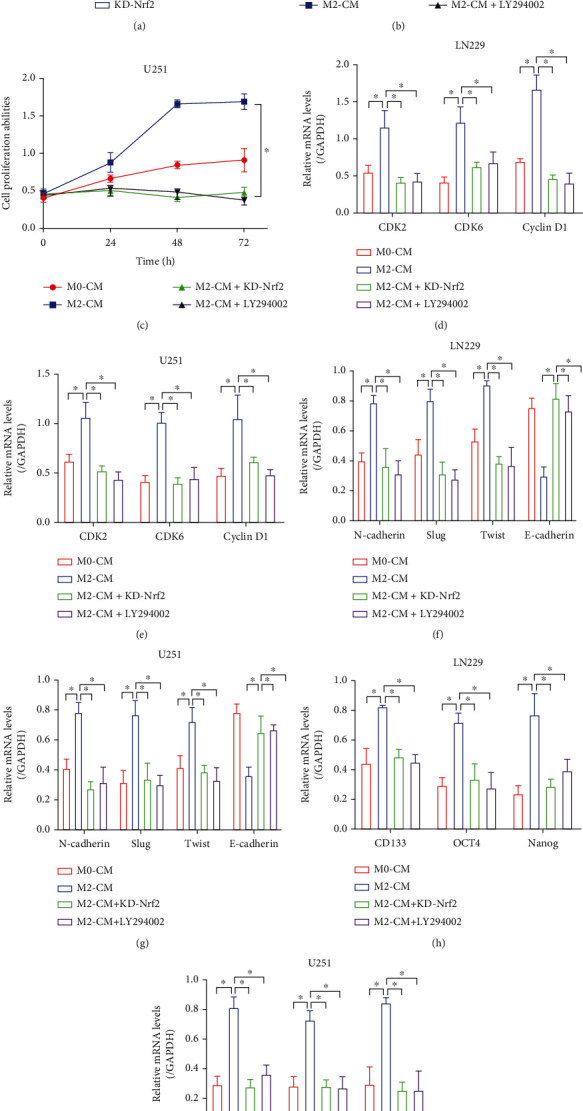
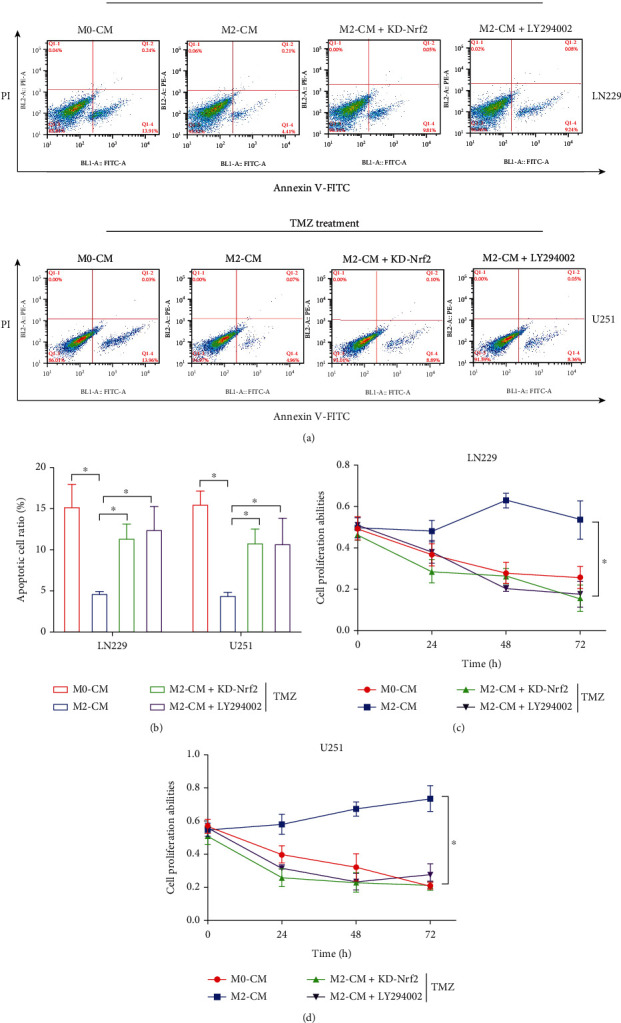
Hypoxia-induced M2 phenotypes of tumor associated macrophages (TAMs) promote the development and chemoresistance of multiple types of cancers, including glioblastoma (GBM). However, the detailed molecular mechanisms have not been fully understood. In this study, we firstly reported that hypoxic pressure promoted M2 macrophage generation, which further promoted cancer progression and temozolomide (TMZ) resistance in GBM through secreting vascular endothelial growth factor (VEGF). Specifically, the clinical data suggested that M2 macrophages were significantly enriched in GBM tissues compared with the adjacent normal tissues, and the following in vitro experiments validated that hypoxic pressure promoted M2-polarized macrophages through upregulating hypoxia-inducible factor-1α (HIF-1α). In addition, hypoxic M2 macrophages VEGF-dependently promoted cell proliferation, epithelial-mesenchymal transition (EMT), glioblastoma stem cell (GSC) properties, and TMZ resistance in GBM cells through activating the PI3K/Akt/Nrf2 pathway. Also, M2 macrophages secreted VEGF to accelerate angiogenesis in human umbilical vein endothelial cells (HUVECs) through interacting with its receptor VEGFR. In general, we concluded that hypoxic M2 macrophages contributed to cancer progression, stemness, drug resistance, and angiogenesis in GBM through secreting VEGF, and our data supported the notion that targeting hypoxia-associated M2 macrophages might be an effective treatment strategy for GBM in clinical practices.
Copyright © 2022 Ge Zhang et al.
Conflict of interest statement
The authors have no conflict of interest.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
