

KRIT1: A Traffic Warden at the Busy Crossroads Between Redox Signaling and the Pathogenesis of Cerebral Cavernous Malformation Disease
- PMID: 36047808
- PMCID: PMC10039281
- DOI: 10.1089/ars.2021.0263
KRIT1: A Traffic Warden at the Busy Crossroads Between Redox Signaling and the Pathogenesis of Cerebral Cavernous Malformation Disease
Abstract
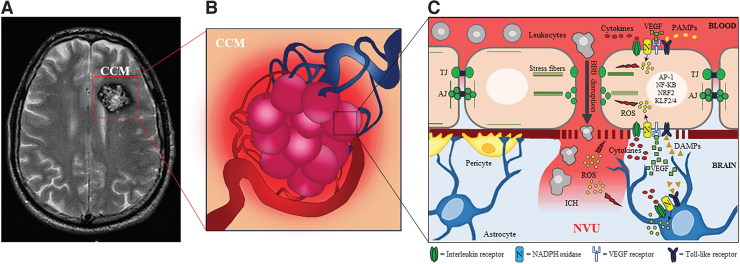
Significance: KRIT1 (Krev interaction trapped 1) is a scaffolding protein that plays a critical role in vascular morphogenesis and homeostasis. Its loss-of-function has been unequivocally associated with the pathogenesis of Cerebral Cavernous Malformation (CCM), a major cerebrovascular disease of genetic origin characterized by defective endothelial cell-cell adhesion and ensuing structural alterations and hyperpermeability in brain capillaries. KRIT1 contributes to the maintenance of endothelial barrier function by stabilizing the integrity of adherens junctions and inhibiting the formation of actin stress fibers. Recent Advances: Among the multiple regulatory mechanisms proposed so far, significant evidence accumulated over the past decade has clearly shown that the role of KRIT1 in the stability of endothelial barriers, including the blood-brain barrier, is largely based on its involvement in the complex machinery governing cellular redox homeostasis and responses to oxidative stress and inflammation. KRIT1 loss-of-function has, indeed, been demonstrated to cause an impairment of major redox-sensitive mechanisms involved in spatiotemporal regulation of cell adhesion and signaling, which ultimately leads to decreased cell-cell junction stability and enhanced sensitivity to oxidative stress and inflammation. Critical Issues: This review explores the redox mechanisms that influence endothelial cell adhesion and barrier function, focusing on the role of KRIT1 in such mechanisms. We propose that this supports a novel model wherein redox signaling forms the common link between the various pathogenetic mechanisms and therapeutic approaches hitherto associated with CCM disease. Future Directions: A comprehensive characterization of the role of KRIT1 in redox control of endothelial barrier physiology and defense against oxy-inflammatory insults will provide valuable insights into the development of precision medicine strategies. Antioxid. Redox Signal. 38, 496-528.
Keywords: GTPases; KRIT1; MAPKs; NADPH oxidases; cerebral cavernous malformation (CCM); cerebrovascular disease; endothelial cell adhesion and barrier function; endothelial-to-mesenchymal transition (EndMT); inflammation; oxidative post-translational modifications (Ox-PTMs); oxidative stress; reactive oxygen species (ROS); redox homeostasis and signaling.
Conflict of interest statement
The authors declare no conflict of interest.
Figures






References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
