

Targeting acetylcholine signaling modulates persistent drug tolerance in EGFR-mutant lung cancer and impedes tumor relapse
- PMID: 36048538
- PMCID: PMC9566900
- DOI: 10.1172/JCI160152
Targeting acetylcholine signaling modulates persistent drug tolerance in EGFR-mutant lung cancer and impedes tumor relapse
Abstract
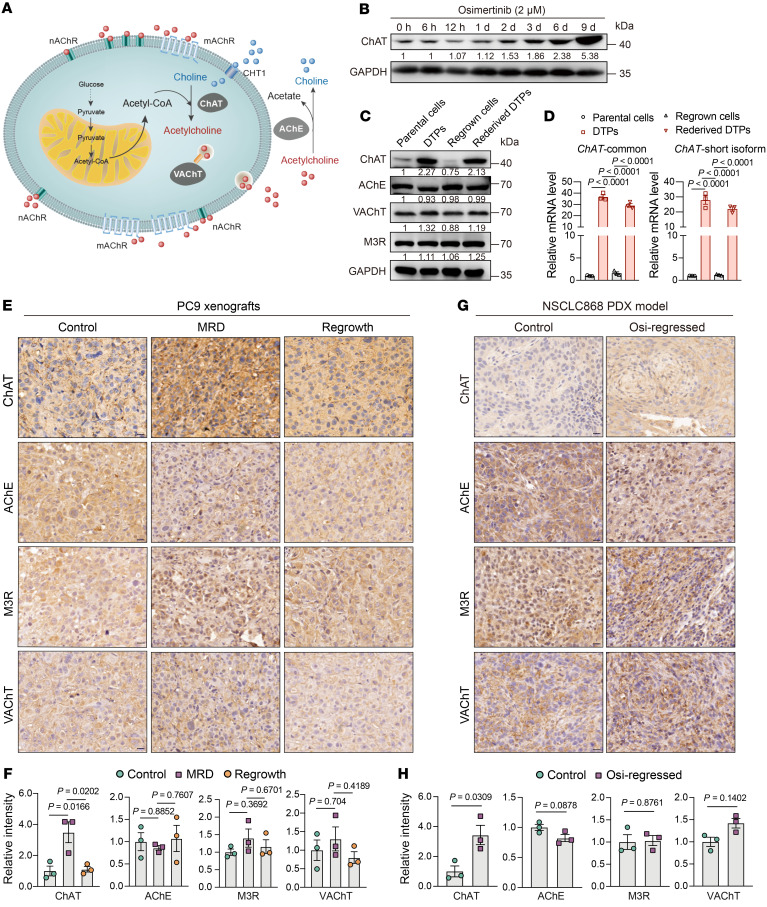
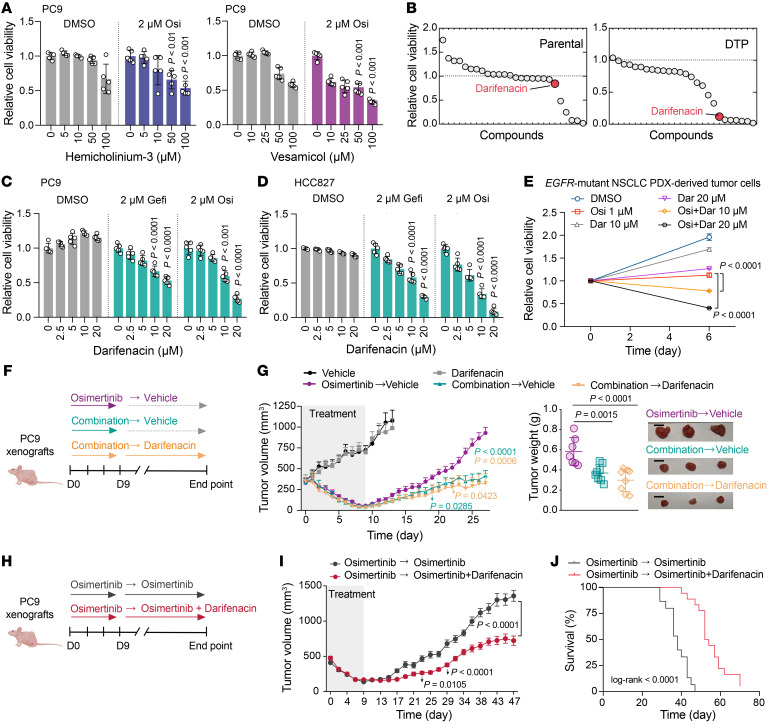
Although first-line epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) therapy is effective for treating EGFR-mutant non-small cell lung cancer (NSCLC), it is now understood that drug-tolerant persister (DTP) cells escaping from initial treatment eventually drives drug resistance. Here, through integration of metabolomics and transcriptomics, we found that the neurotransmitter acetylcholine (ACh) was specifically accumulated in DTP cells, and demonstrated that treatment with EGFR-TKI heightened the expression of the rate-limiting enzyme choline acetyltransferase (ChAT) in ACh biosynthesis via YAP mediation. Genetic and pharmacological manipulation of ACh biosynthesis or ACh signaling could predictably regulate the extent of DTP formation in vitro and in vivo. Strikingly, pharmacologically targeting ACh/M3R signaling with an FDA-approved drug, darifenacin, retarded tumor relapse in vivo. Mechanistically, upregulated ACh metabolism mediated drug tolerance in part through activating WNT signaling via ACh muscarinic receptor 3 (M3R). Importantly, we showed that aberrant ACh metabolism in patients with NSCLC played a potential role in predicting EGFR-TKI response rate and progression-free survival. Our study therefore defines a therapeutic strategy - targeting the ACh/M3R/WNT axis - for manipulating EGFR TKI drug tolerance in the treatment of NSCLC.
Keywords: Drug therapy; Lung cancer; Metabolism; Oncology; Tolerance.
Figures






References
-
- Boumahdi S, de Sauvage FJ. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov. 2019;19(1):39–56. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
