

Cell type-specific inference of differential expression in spatial transcriptomics
- PMID: 36050488
- PMCID: PMC10463137
- DOI: 10.1038/s41592-022-01575-3
Cell type-specific inference of differential expression in spatial transcriptomics
Abstract
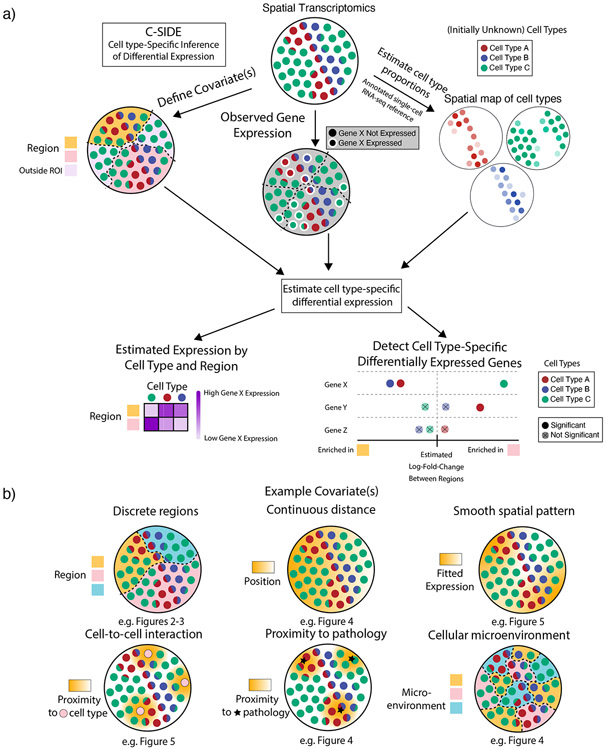
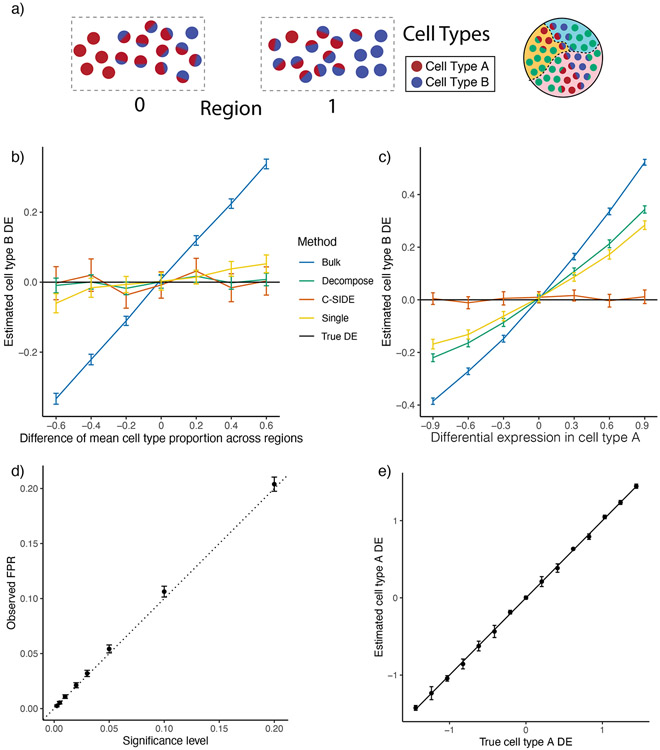
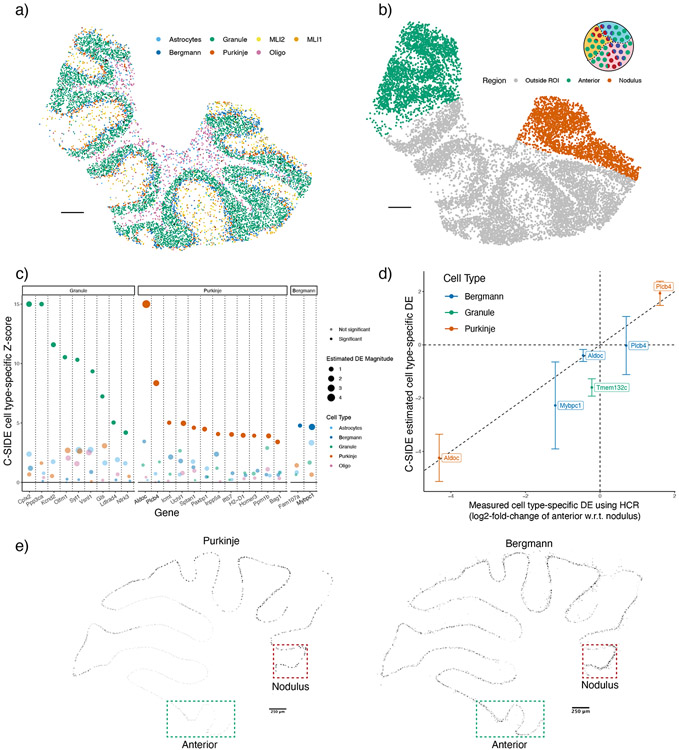
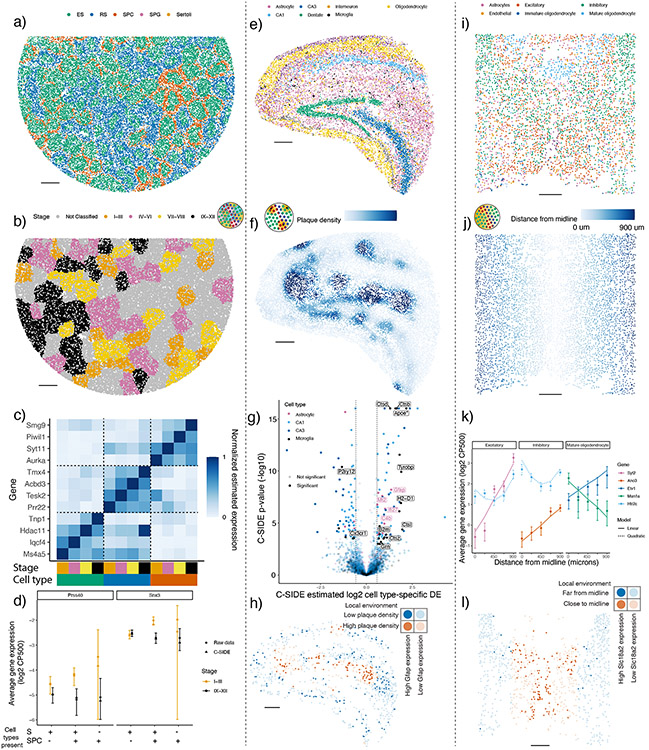
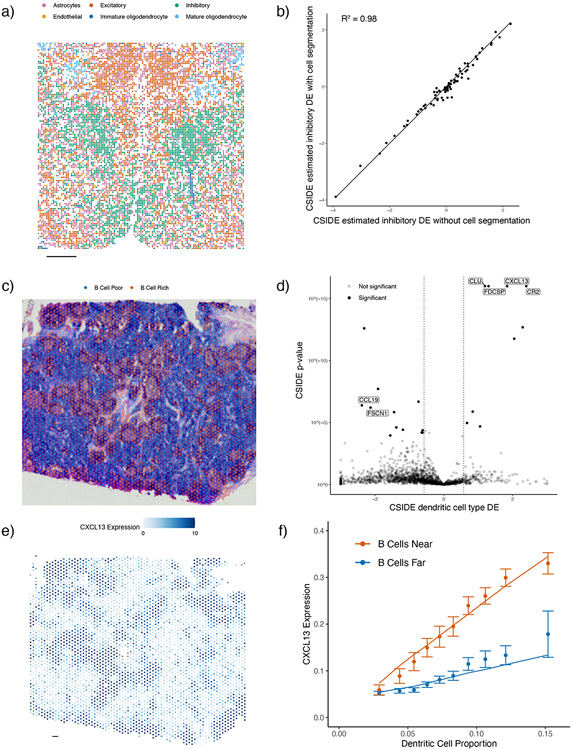
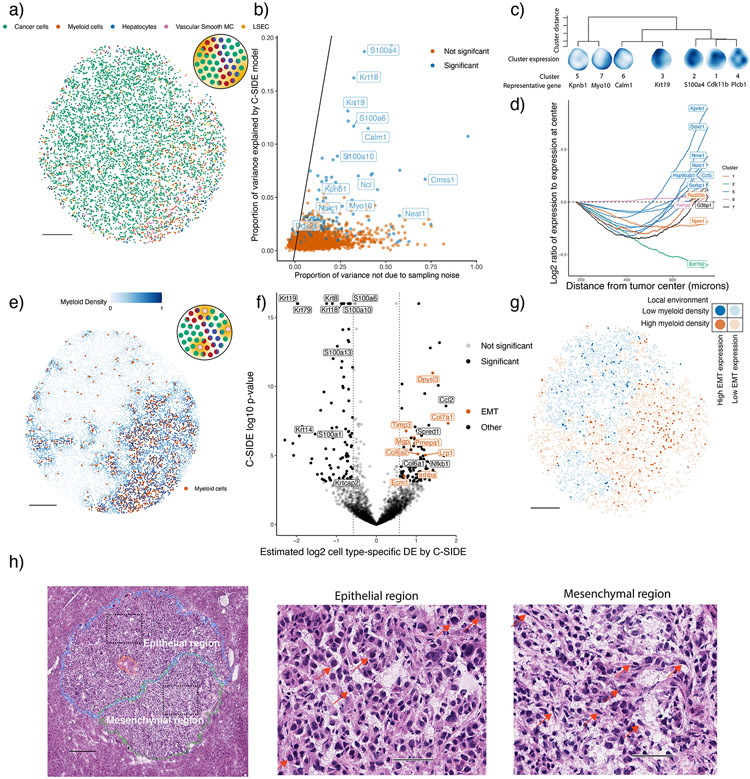
A central problem in spatial transcriptomics is detecting differentially expressed (DE) genes within cell types across tissue context. Challenges to learning DE include changing cell type composition across space and measurement pixels detecting transcripts from multiple cell types. Here, we introduce a statistical method, cell type-specific inference of differential expression (C-SIDE), that identifies cell type-specific DE in spatial transcriptomics, accounting for localization of other cell types. We model gene expression as an additive mixture across cell types of log-linear cell type-specific expression functions. C-SIDE's framework applies to many contexts: DE due to pathology, anatomical regions, cell-to-cell interactions and cellular microenvironment. Furthermore, C-SIDE enables statistical inference across multiple/replicates. Simulations and validation experiments on Slide-seq, MERFISH and Visium datasets demonstrate that C-SIDE accurately identifies DE with valid uncertainty quantification. Last, we apply C-SIDE to identify plaque-dependent immune activity in Alzheimer's disease and cellular interactions between tumor and immune cells. We distribute C-SIDE within the R package https://github.com/dmcable/spacexr .
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
Similar articles
-
Robust decomposition of cell type mixtures in spatial transcriptomics.Nat Biotechnol. 2022 Apr;40(4):517-526. doi: 10.1038/s41587-021-00830-w. Epub 2021 Feb 18. Nat Biotechnol. 2022. PMID: 33603203 Free PMC article.
-
stVAE deconvolves cell-type composition in large-scale cellular resolution spatial transcriptomics.Bioinformatics. 2023 Oct 3;39(10):btad642. doi: 10.1093/bioinformatics/btad642. Bioinformatics. 2023. PMID: 37862237 Free PMC article.
-
Reference-free cell type deconvolution of multi-cellular pixel-resolution spatially resolved transcriptomics data.Nat Commun. 2022 Apr 29;13(1):2339. doi: 10.1038/s41467-022-30033-z. Nat Commun. 2022. PMID: 35487922 Free PMC article.
-
Computational solutions for spatial transcriptomics.Comput Struct Biotechnol J. 2022 Sep 1;20:4870-4884. doi: 10.1016/j.csbj.2022.08.043. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 36147664 Free PMC article. Review.
-
Spatial transcriptomics: Technologies, applications and experimental considerations.Genomics. 2023 Sep;115(5):110671. doi: 10.1016/j.ygeno.2023.110671. Epub 2023 Jun 21. Genomics. 2023. PMID: 37353093 Free PMC article. Review.
Cited by
-
Identifying spatially variable genes by projecting to morphologically relevant curves.bioRxiv [Preprint]. 2024 Nov 21:2024.11.21.624653. doi: 10.1101/2024.11.21.624653. bioRxiv. 2024. PMID: 39605709 Free PMC article. Preprint.
-
Scalable imaging-free spatial genomics through computational reconstruction.bioRxiv [Preprint]. 2024 Sep 16:2024.08.05.606465. doi: 10.1101/2024.08.05.606465. bioRxiv. 2024. PMID: 39149311 Free PMC article. Preprint.
-
DHODH modulates immune evasion of cancer cells via CDP-Choline dependent regulation of phospholipid metabolism and ferroptosis.Nat Commun. 2025 Apr 24;16(1):3867. doi: 10.1038/s41467-025-59307-y. Nat Commun. 2025. PMID: 40274823 Free PMC article.
-
Answering open questions in biology using spatial genomics and structured methods.BMC Bioinformatics. 2024 Sep 4;25(1):291. doi: 10.1186/s12859-024-05912-5. BMC Bioinformatics. 2024. PMID: 39232666 Free PMC article. Review.
-
Spatial maps of T cell receptors and transcriptomes reveal distinct immune niches and interactions in the adaptive immune response.Immunity. 2022 Oct 11;55(10):1940-1952.e5. doi: 10.1016/j.immuni.2022.09.002. Immunity. 2022. PMID: 36223726 Free PMC article.
References
-
- 10x Genomics. 10x genomics: Visium spatial gene expression. https://www.10xgenomics.com/solutions/spatial-gene-expression/ (2020).
Methods References
-
- Yuan YX A review of trust region algorithms for optimization. In Iciam, vol. 99, 271–282 (2000).
-
- Van der Vaart AW Asymptotic statistics, vol. 3 (Cambridge university press, 2000).
-
- Benjamini Y & Hochberg Y Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal statistical society: series B (Methodological) 57, 289–300 (1995).
-
- DerSimonian R & Laird N Meta-analysis in clinical trials. Controlled clinical trials 7, 177–188 (1986). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
