

The genomic landscape of pediatric acute lymphoblastic leukemia
- PMID: 36050548
- PMCID: PMC9700506
- DOI: 10.1038/s41588-022-01159-z
The genomic landscape of pediatric acute lymphoblastic leukemia
Abstract
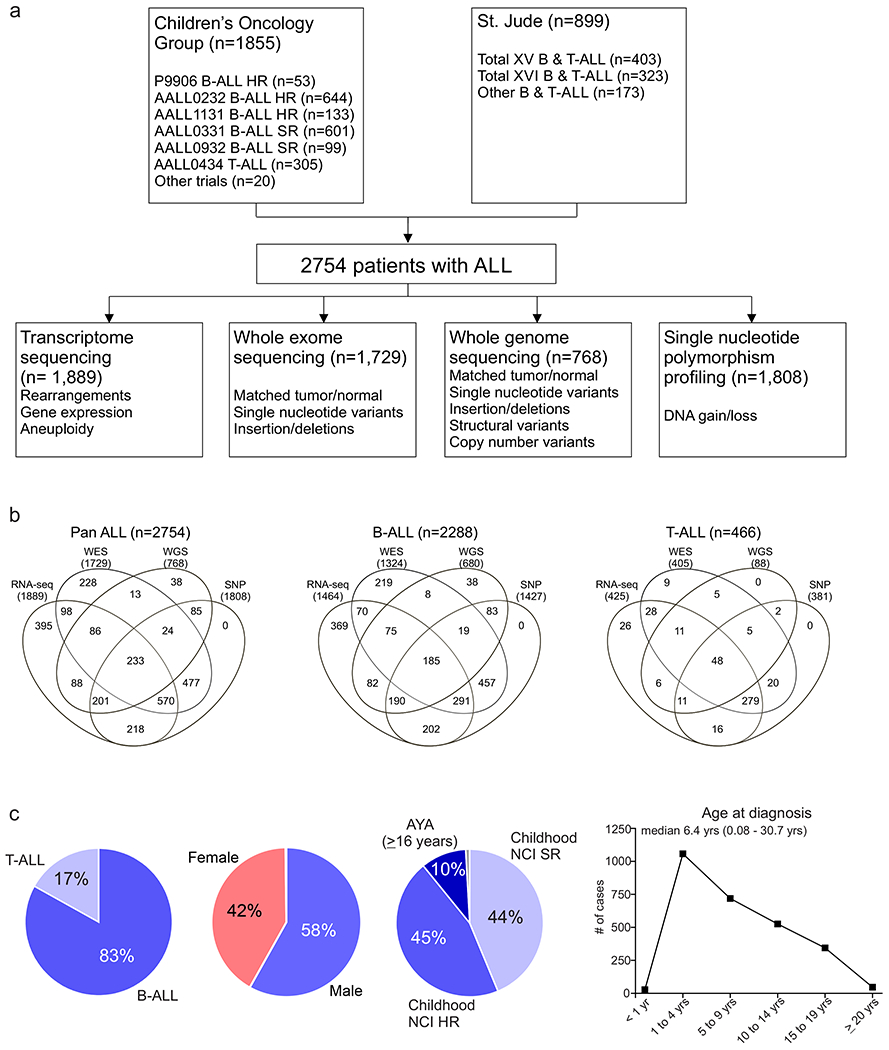
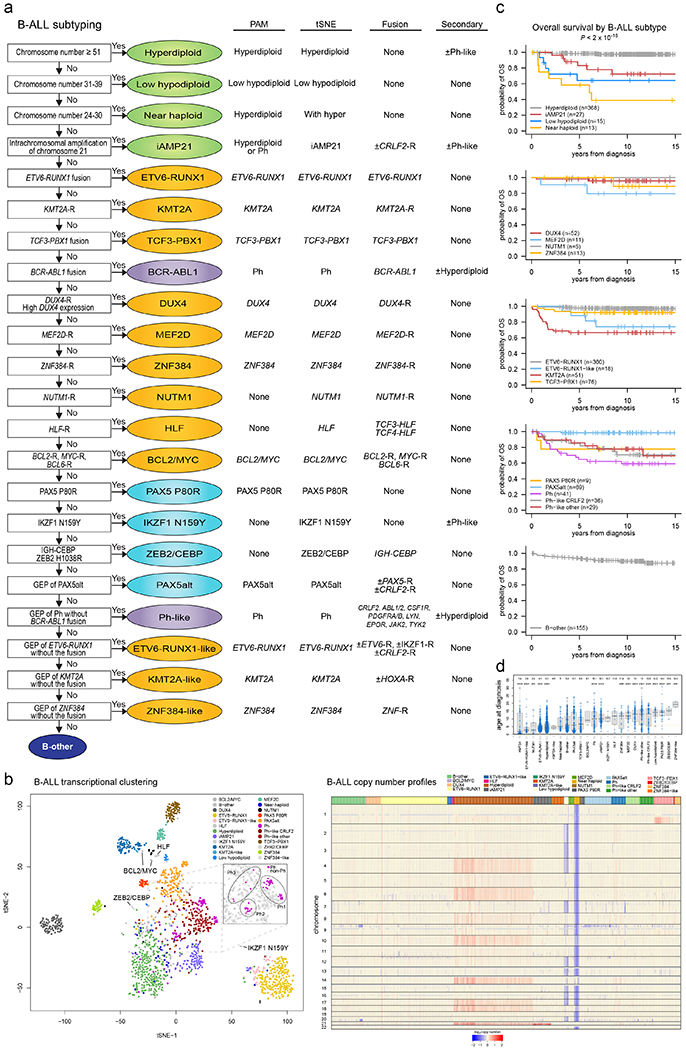
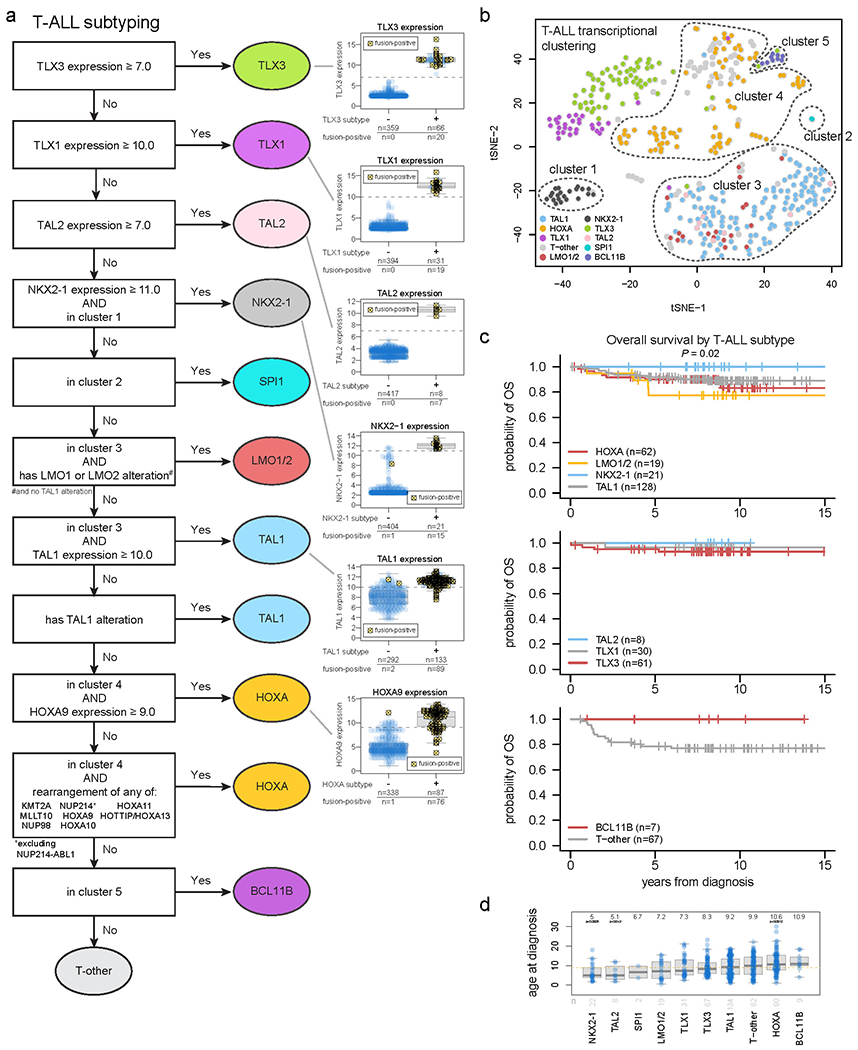
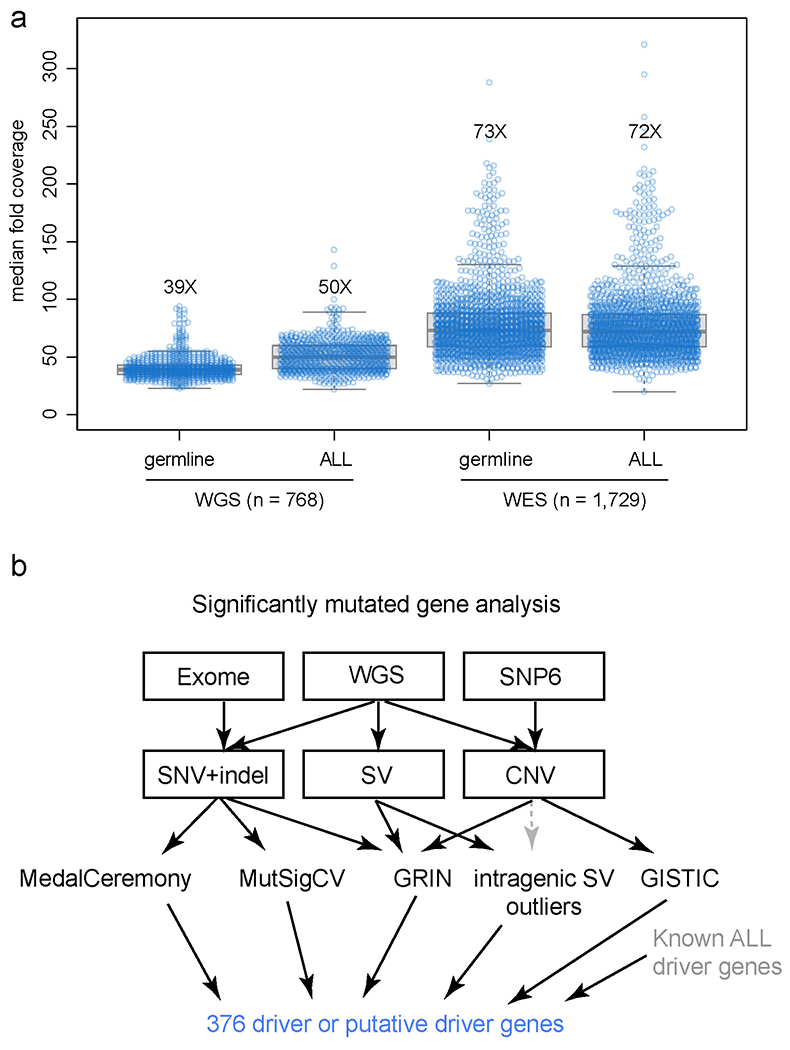
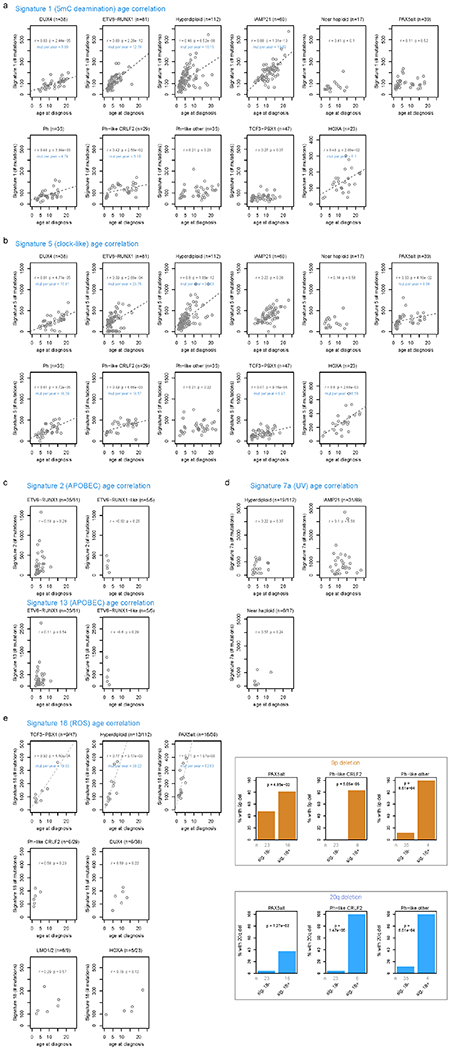
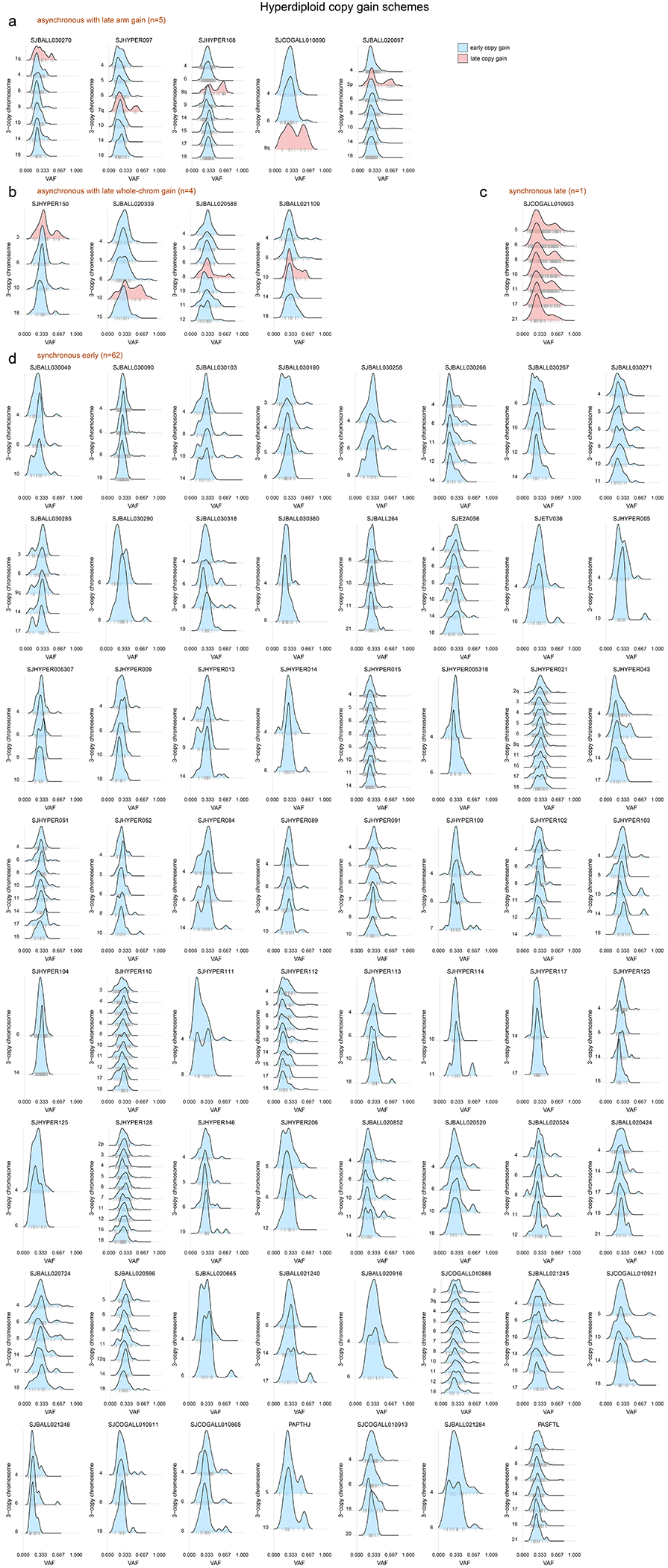
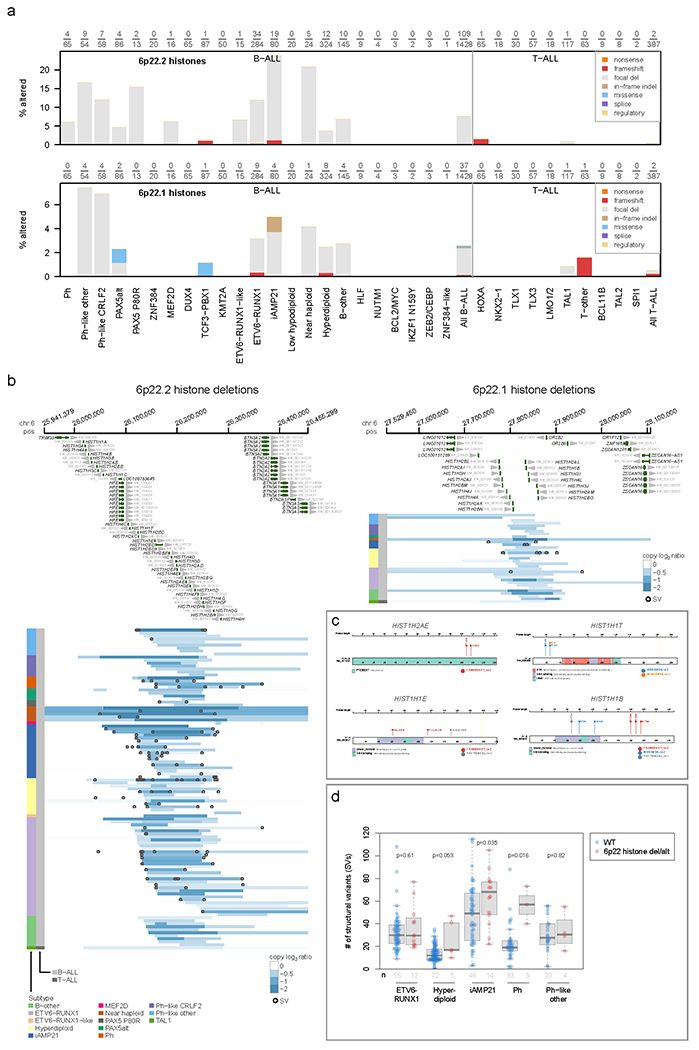
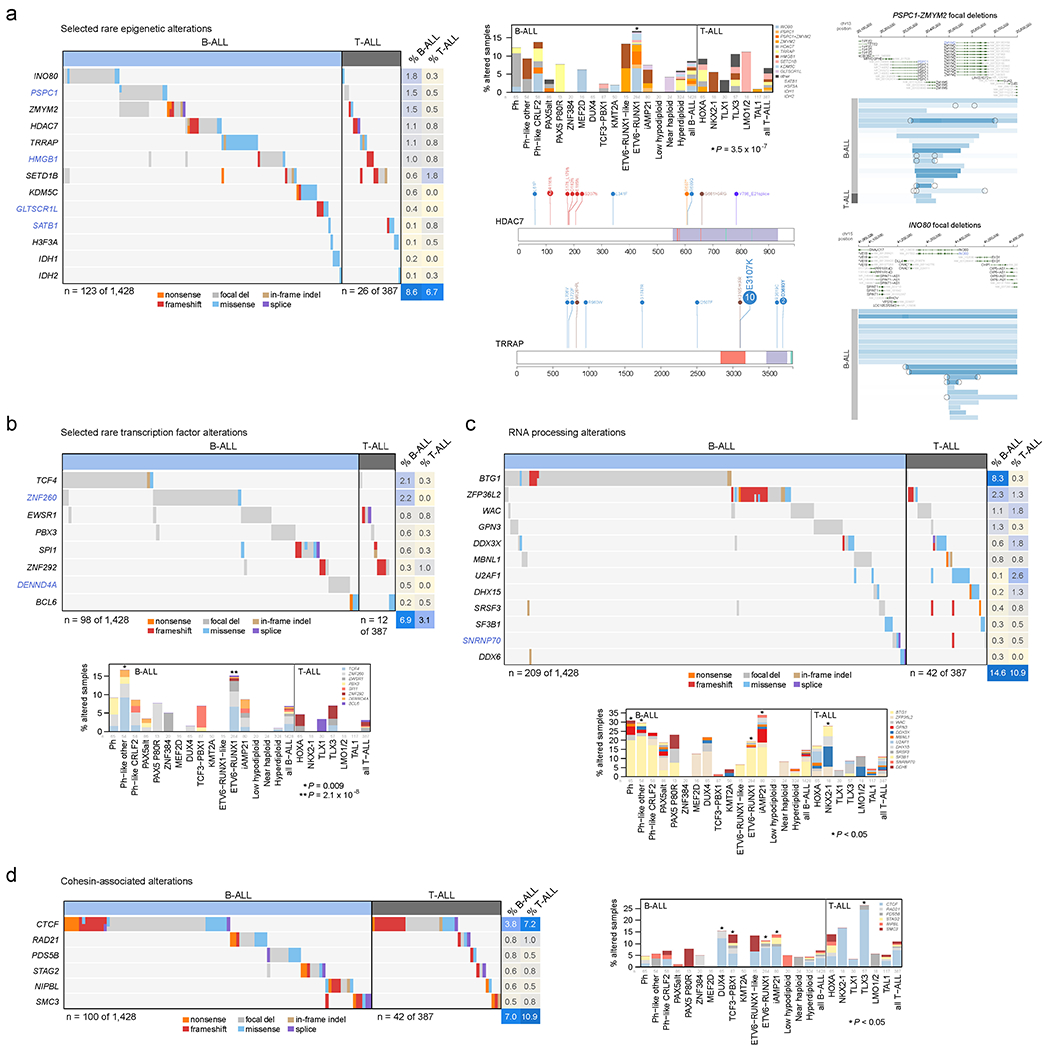
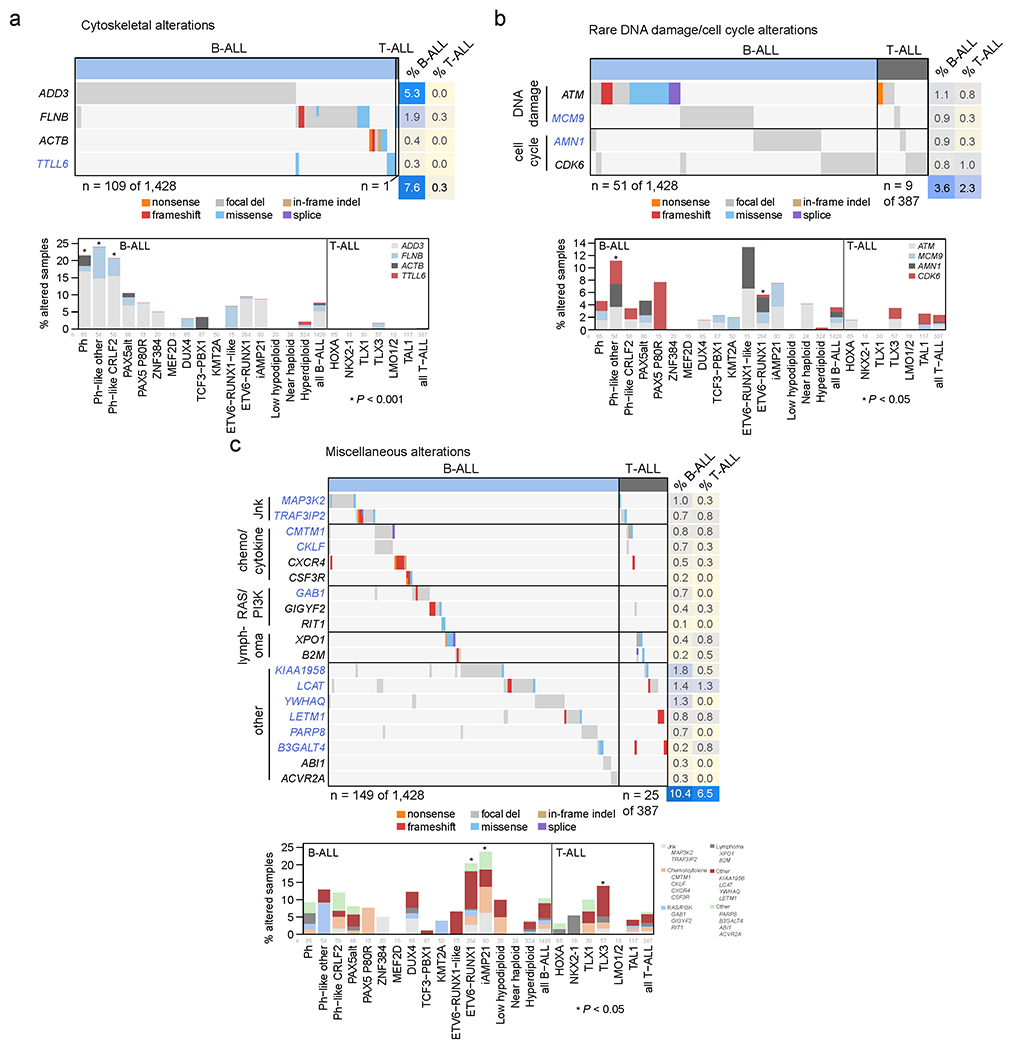
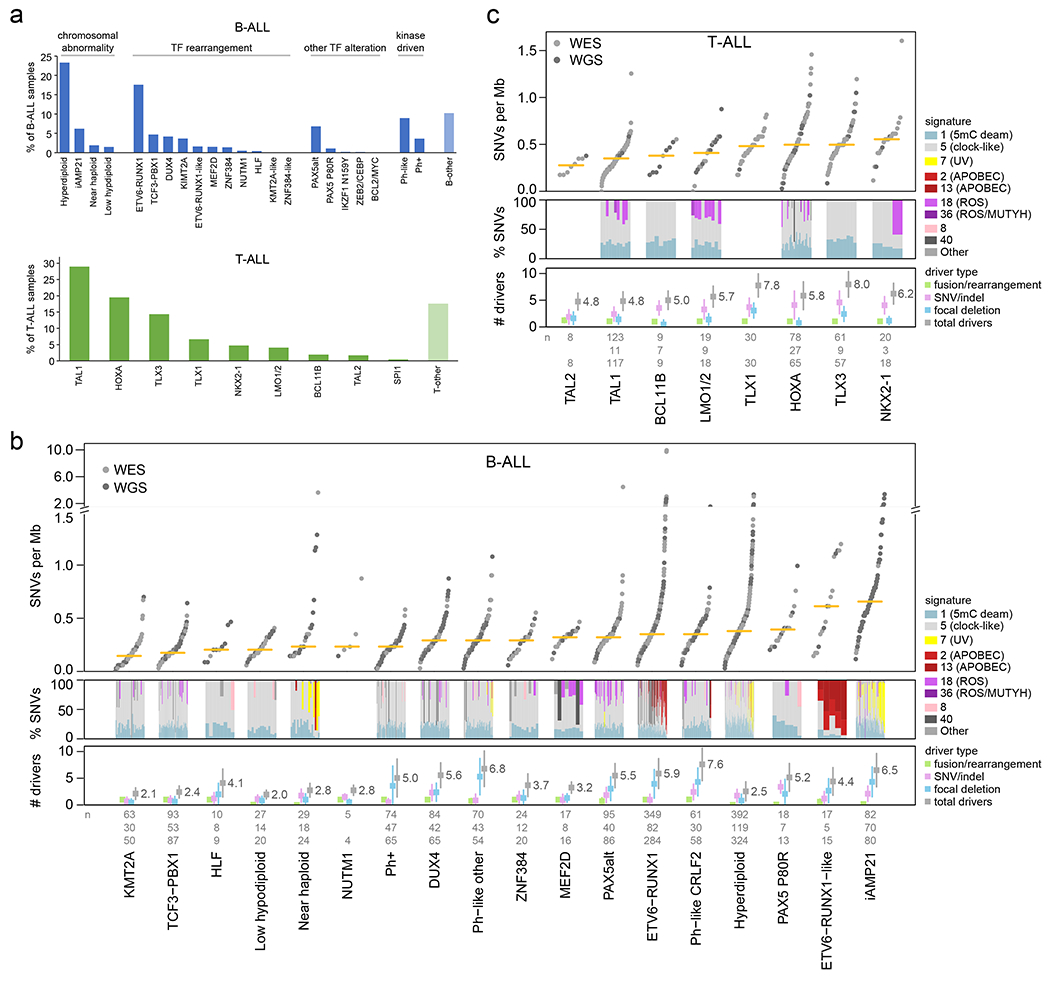
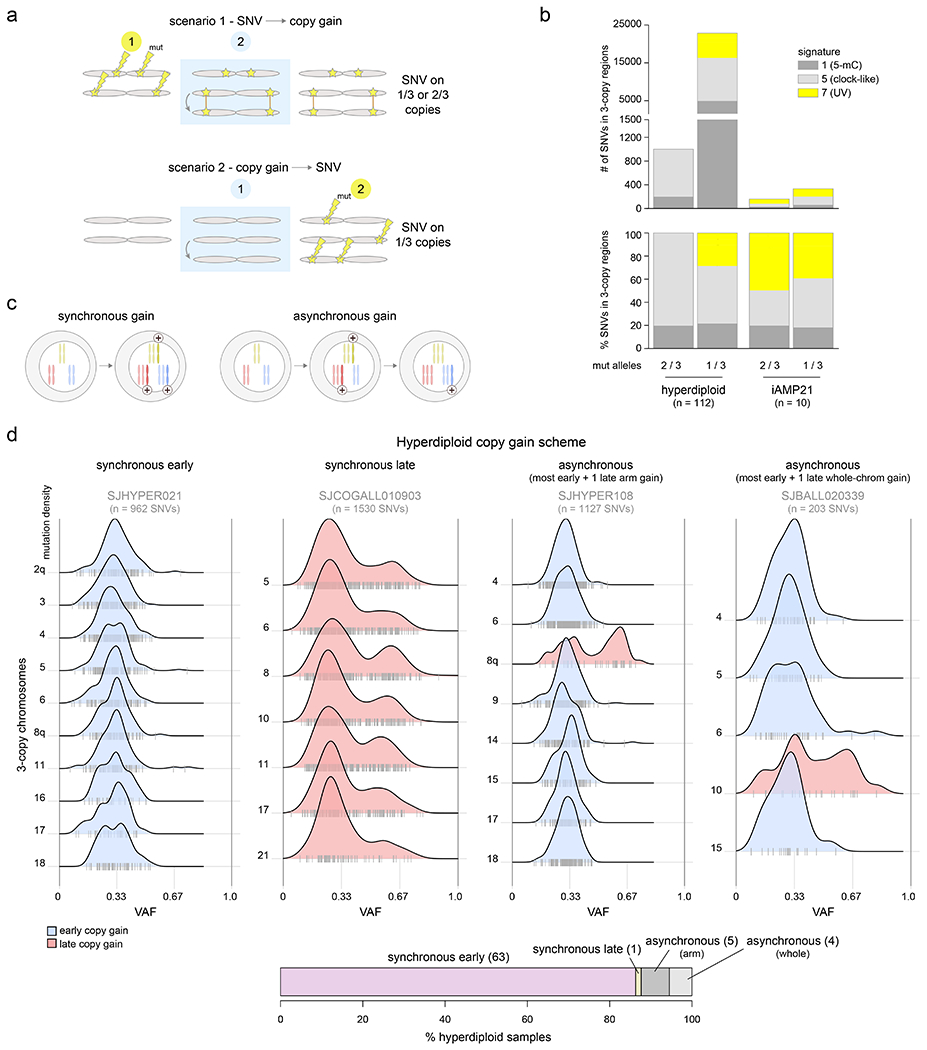
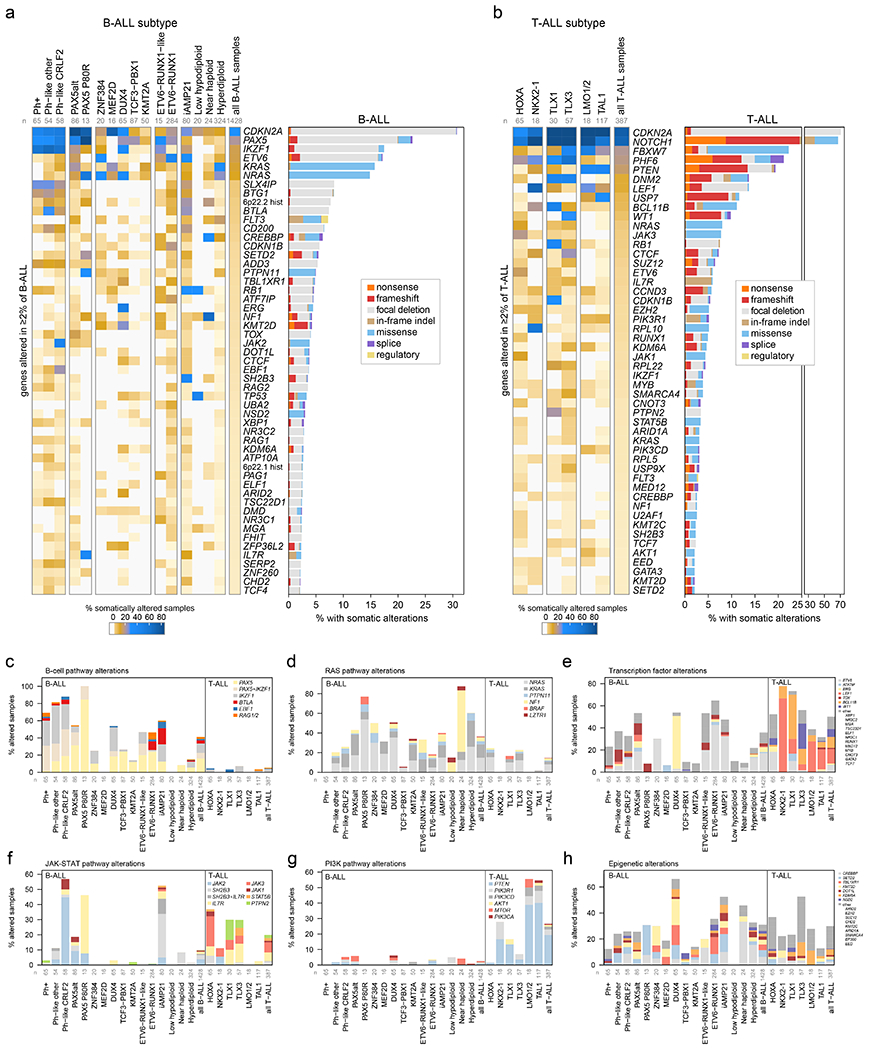
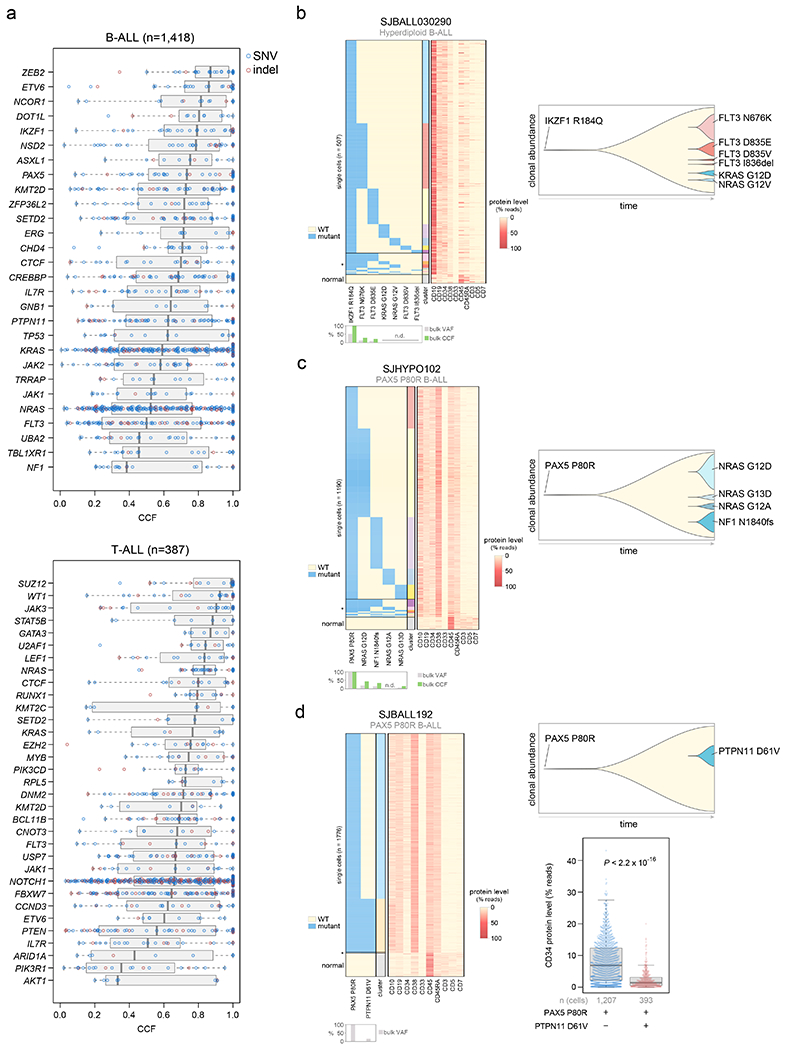
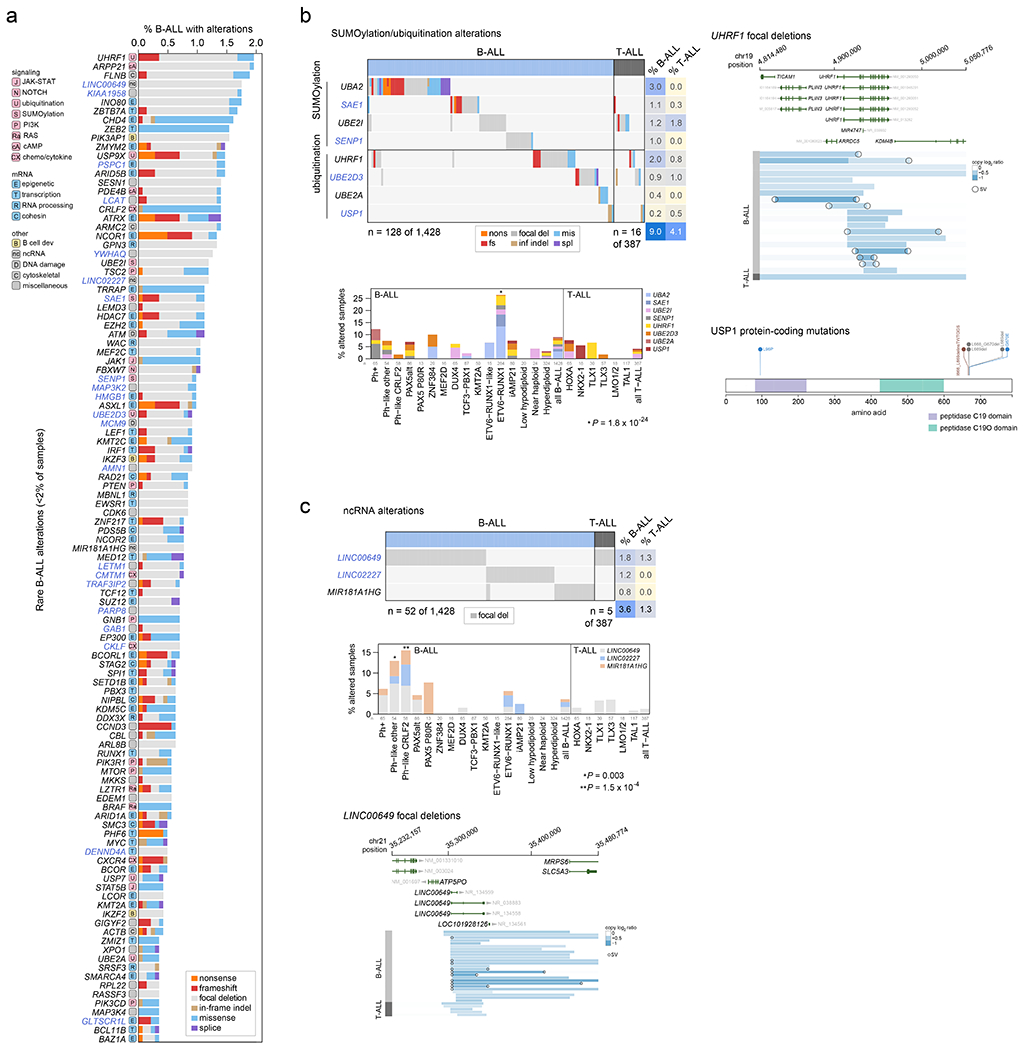
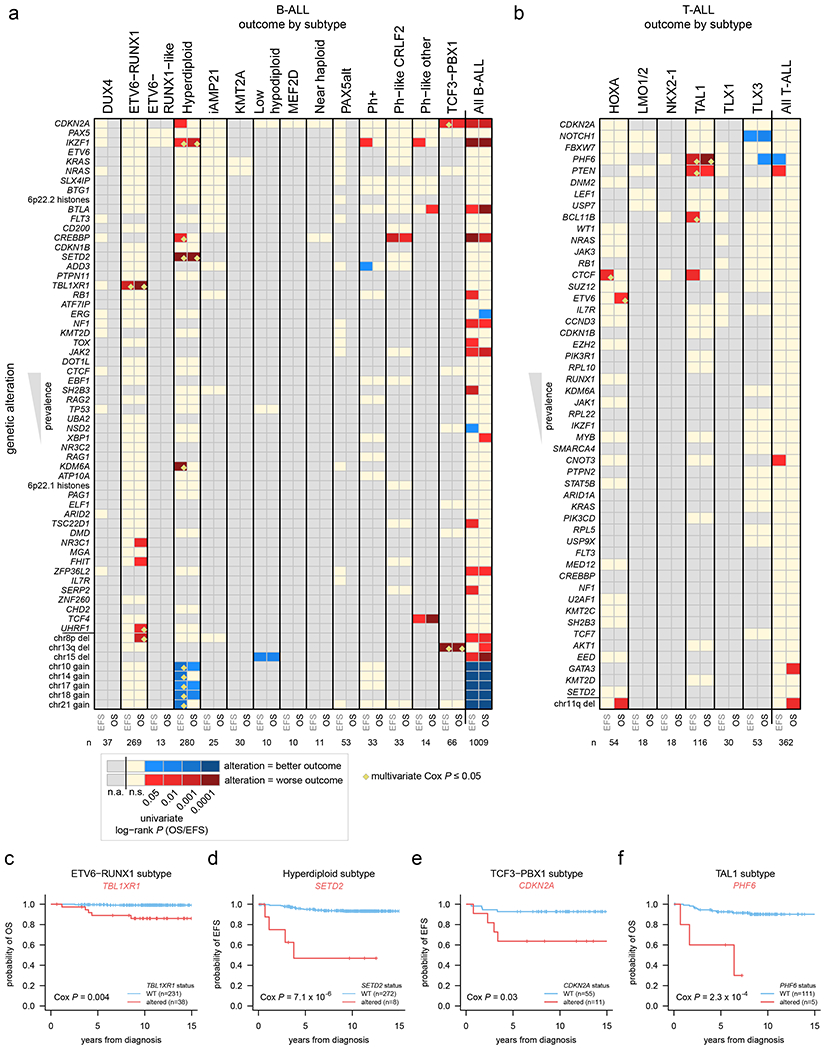
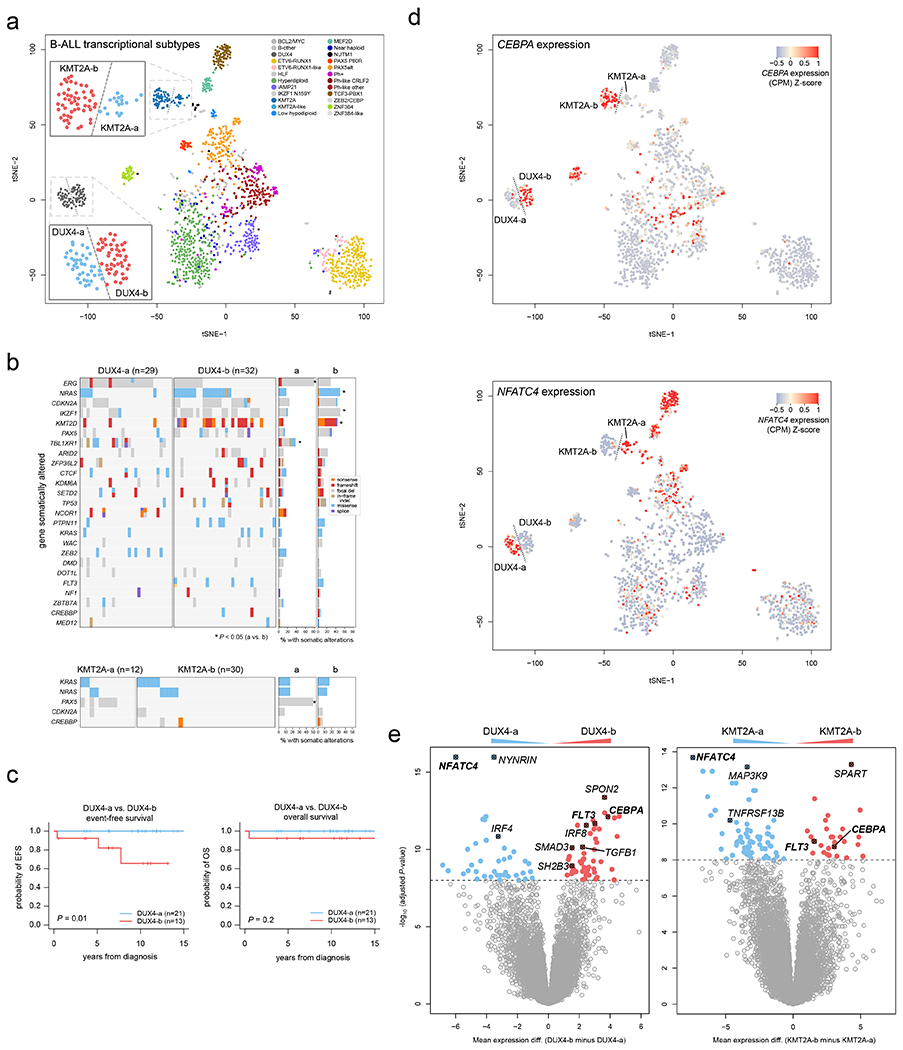
Acute lymphoblastic leukemia (ALL) is the most common childhood cancer. Here, using whole-genome, exome and transcriptome sequencing of 2,754 childhood patients with ALL, we find that, despite a generally low mutation burden, ALL cases harbor a median of four putative somatic driver alterations per sample, with 376 putative driver genes identified varying in prevalence across ALL subtypes. Most samples harbor at least one rare gene alteration, including 70 putative cancer driver genes associated with ubiquitination, SUMOylation, noncoding transcripts and other functions. In hyperdiploid B-ALL, chromosomal gains are acquired early and synchronously before ultraviolet-induced mutation. By contrast, ultraviolet-induced mutations precede chromosomal gains in B-ALL cases with intrachromosomal amplification of chromosome 21. We also demonstrate the prognostic significance of genetic alterations within subtypes. Intriguingly, DUX4- and KMT2A-rearranged subtypes separate into CEBPA/FLT3- or NFATC4-expressing subgroups with potential clinical implications. Together, these results deepen understanding of the ALL genomic landscape and associated outcomes.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures

References
Main References
-
- Roberts KG Why and how to treat Ph-like ALL? Best Pract Res Clin Haematol 31, 351–356 (2018). - PubMed
Methods References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
