

Polygenic enrichment distinguishes disease associations of individual cells in single-cell RNA-seq data
- PMID: 36050550
- PMCID: PMC9891382
- DOI: 10.1038/s41588-022-01167-z
Polygenic enrichment distinguishes disease associations of individual cells in single-cell RNA-seq data
Abstract
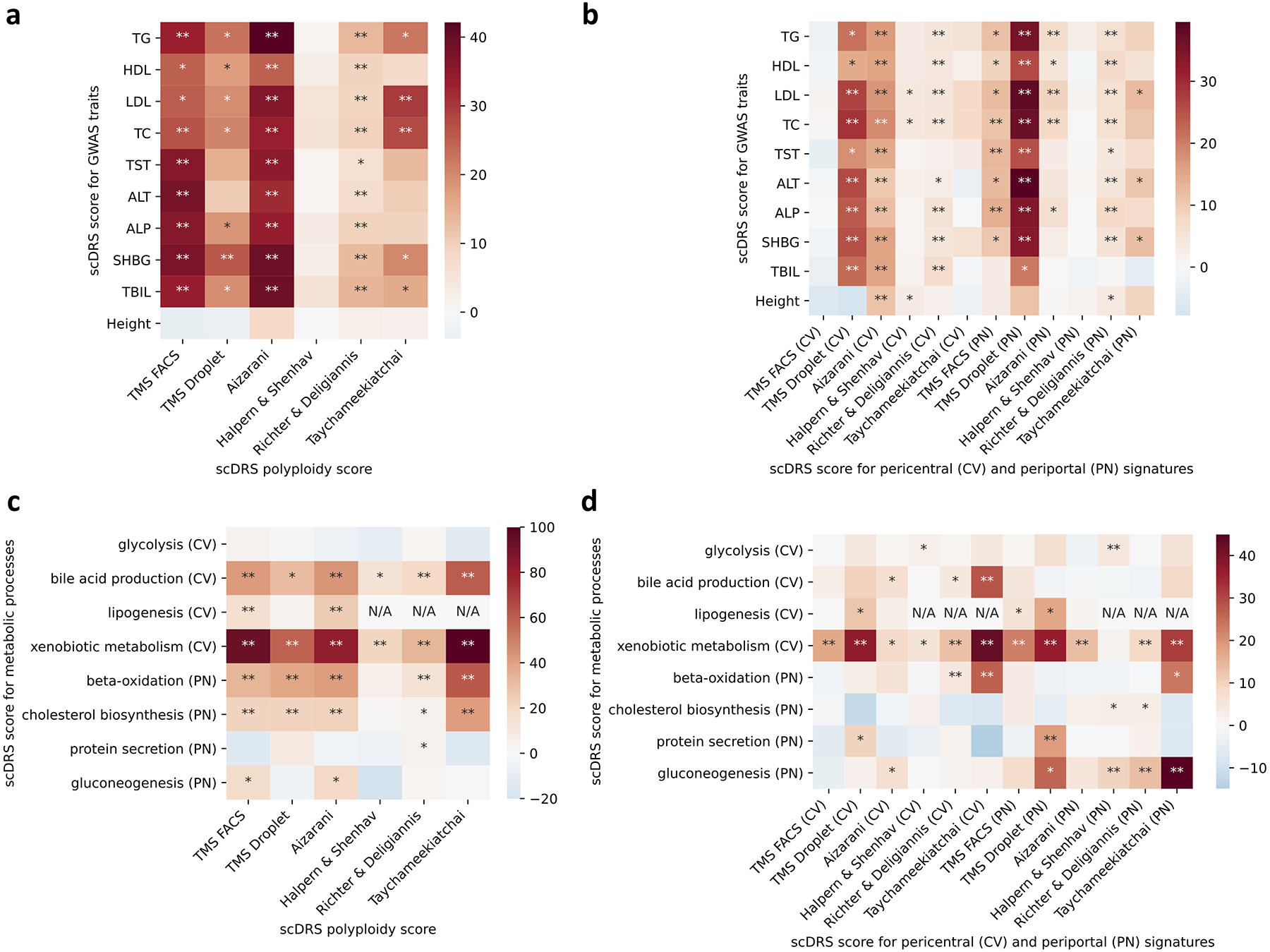
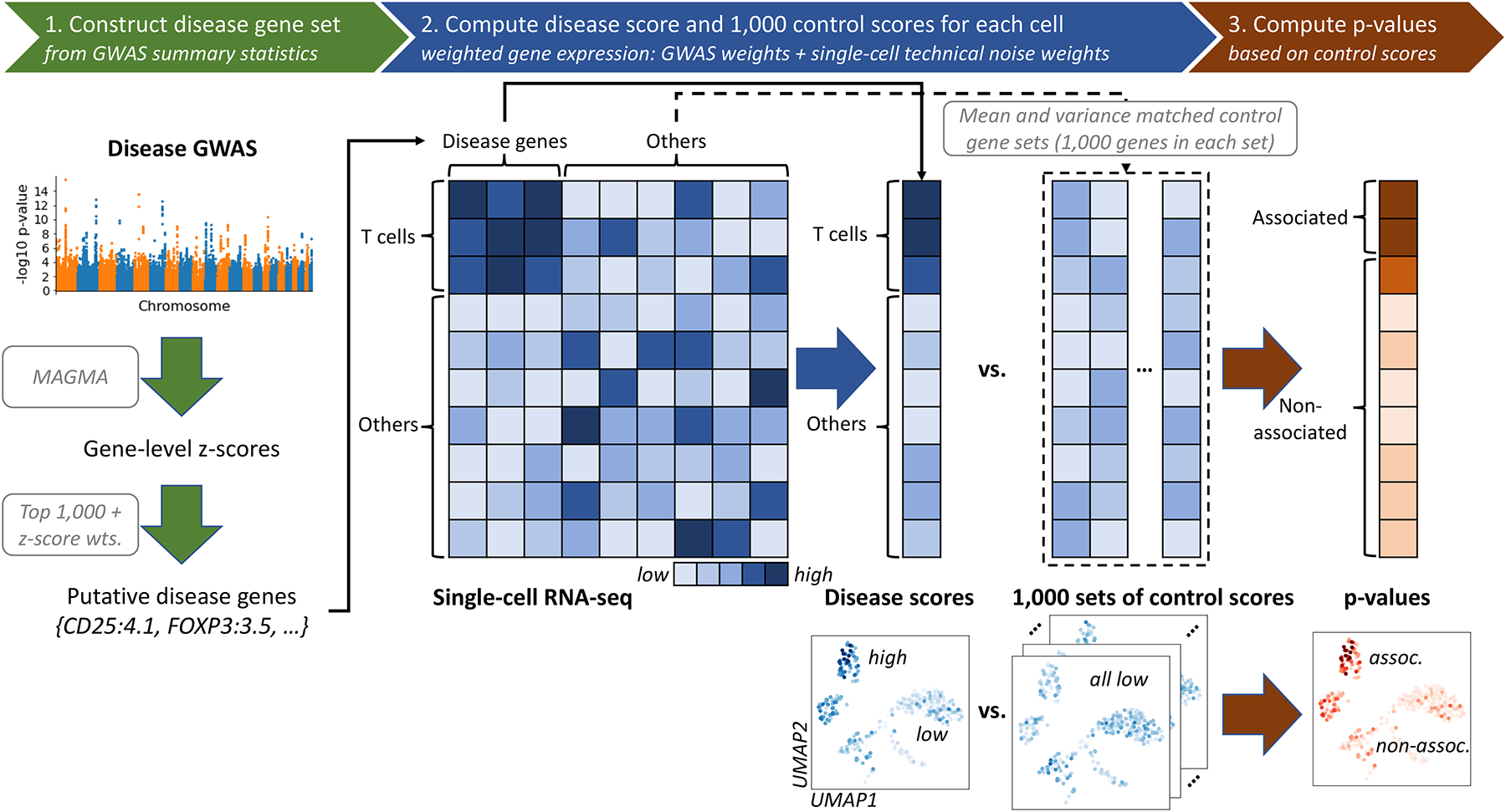
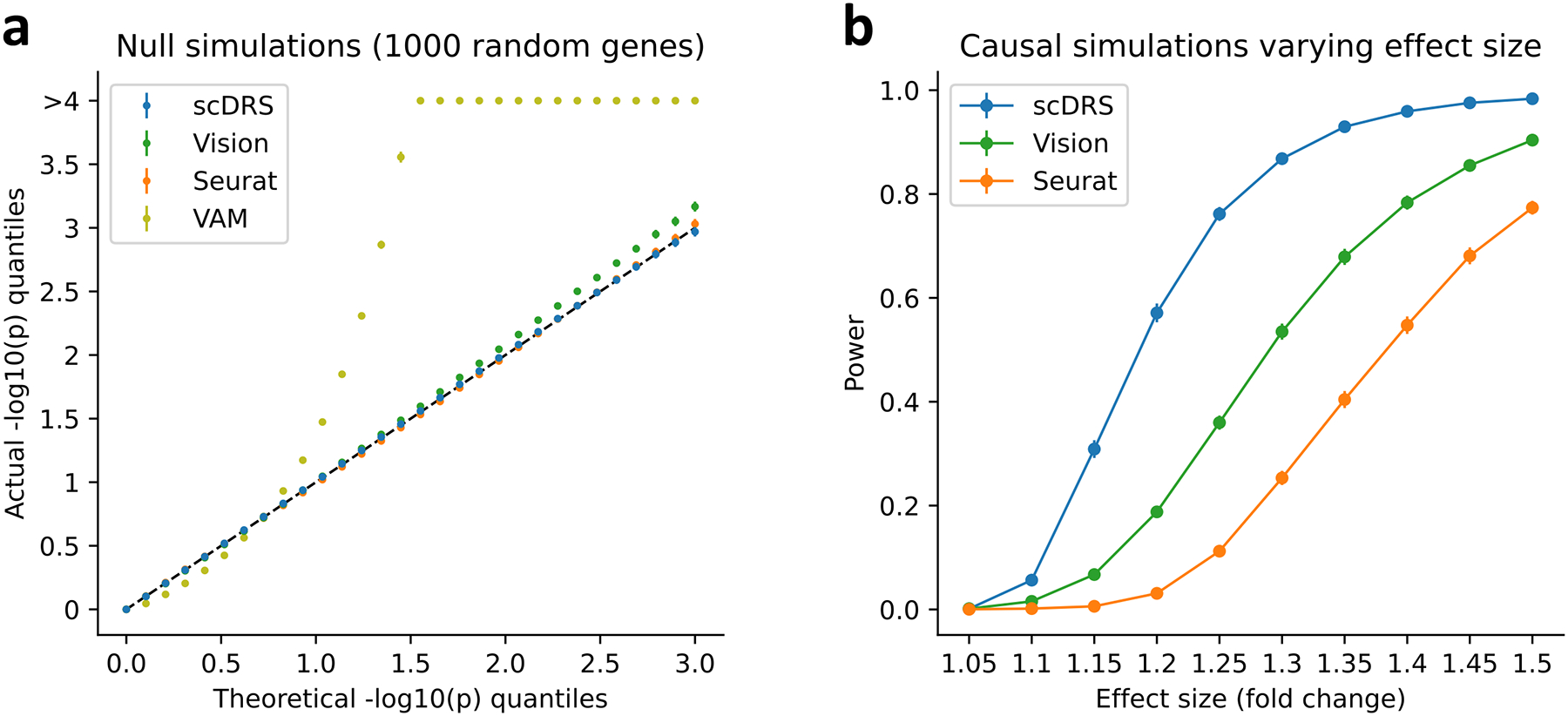
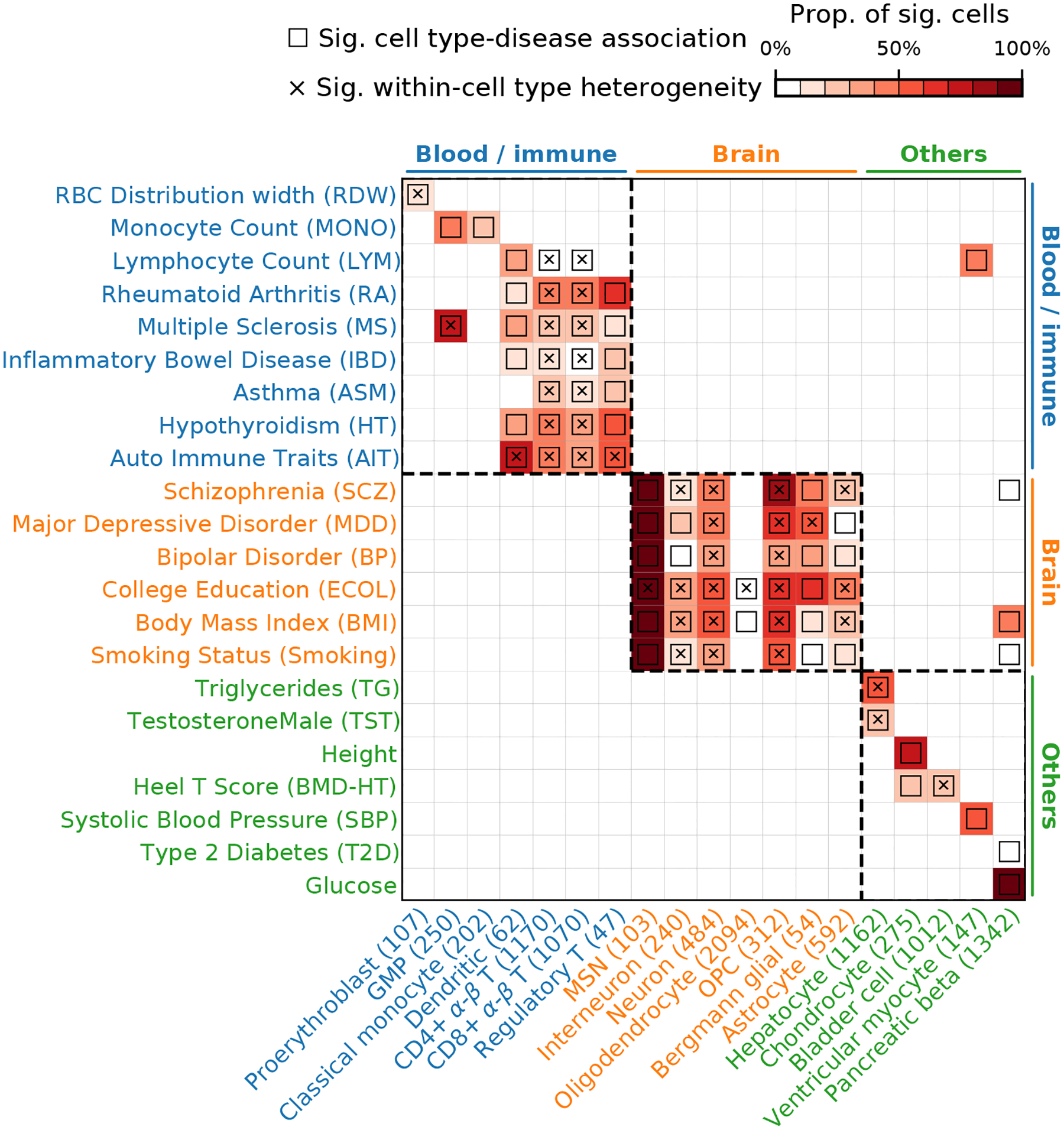
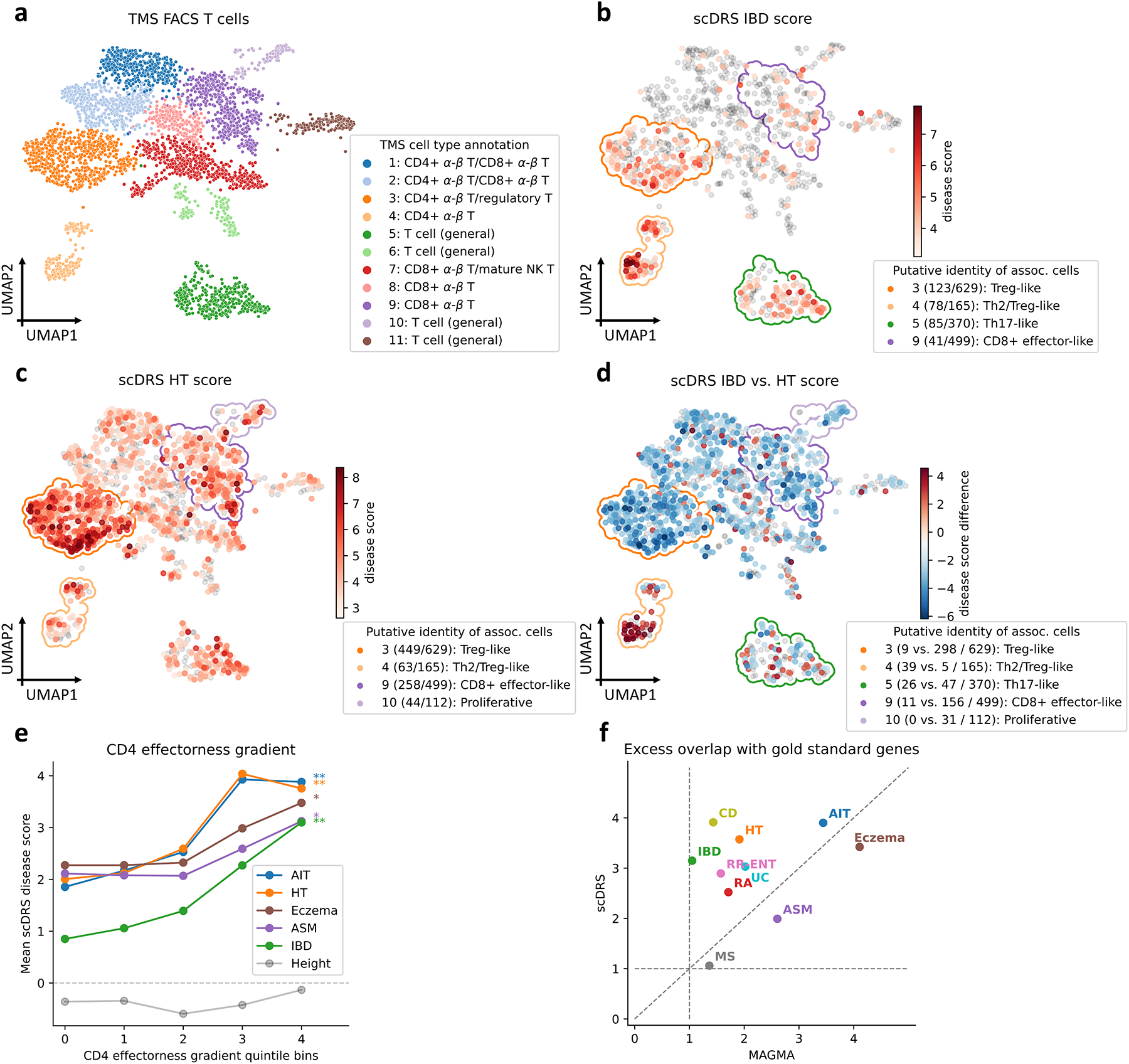
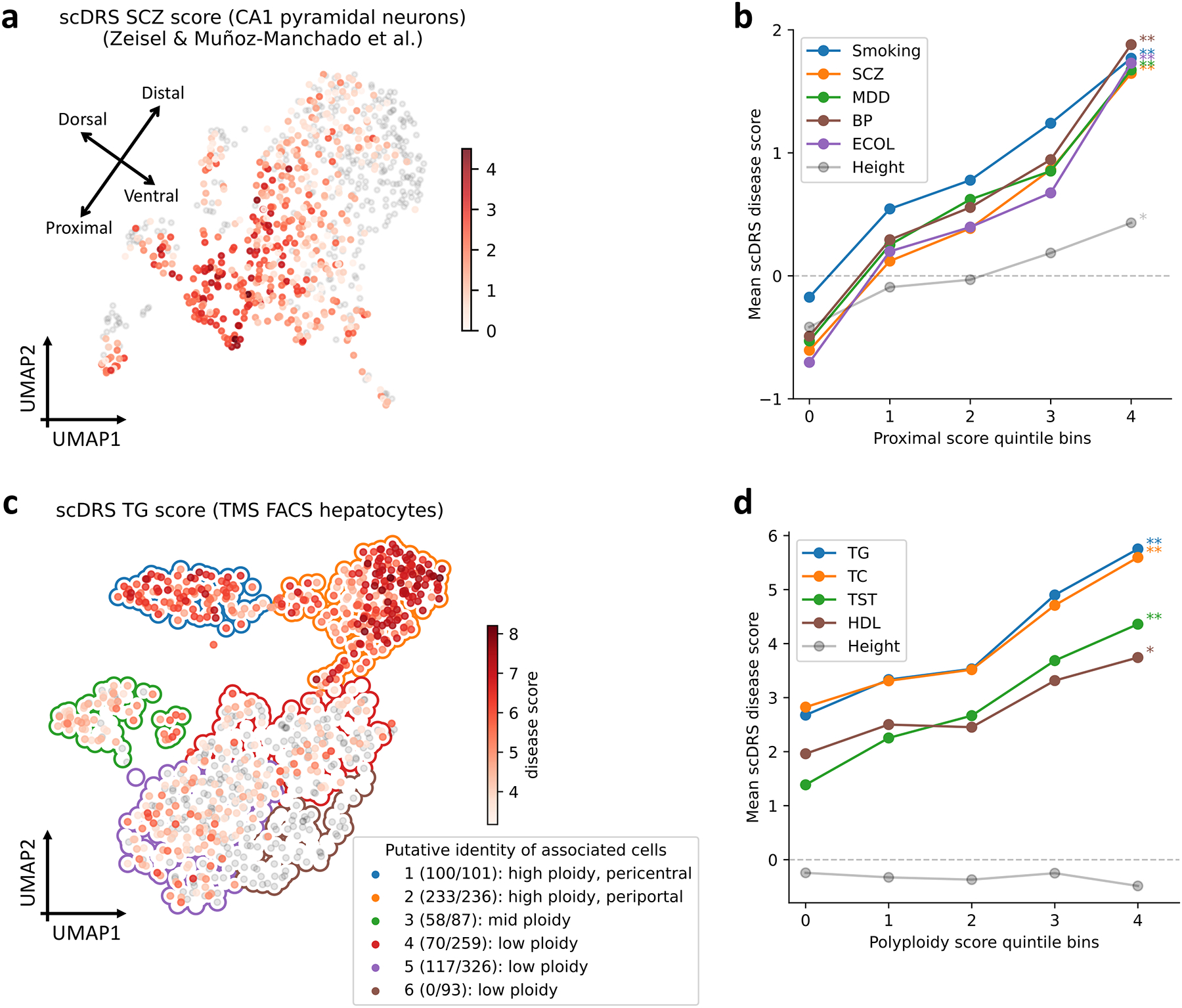
Single-cell RNA sequencing (scRNA-seq) provides unique insights into the pathology and cellular origin of disease. We introduce single-cell disease relevance score (scDRS), an approach that links scRNA-seq with polygenic disease risk at single-cell resolution, independent of annotated cell types. scDRS identifies cells exhibiting excess expression across disease-associated genes implicated by genome-wide association studies (GWASs). We applied scDRS to 74 diseases/traits and 1.3 million single-cell gene-expression profiles across 31 tissues/organs. Cell-type-level results broadly recapitulated known cell-type-disease associations. Individual-cell-level results identified subpopulations of disease-associated cells not captured by existing cell-type labels, including T cell subpopulations associated with inflammatory bowel disease, partially characterized by their effector-like states; neuron subpopulations associated with schizophrenia, partially characterized by their spatial locations; and hepatocyte subpopulations associated with triglyceride levels, partially characterized by their higher ploidy levels. Genes whose expression was correlated with the scDRS score across cells (reflecting coexpression with GWAS disease-associated genes) were strongly enriched for gold-standard drug target and Mendelian disease genes.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing Interests Statement
The authors declare no competing interests.
Figures
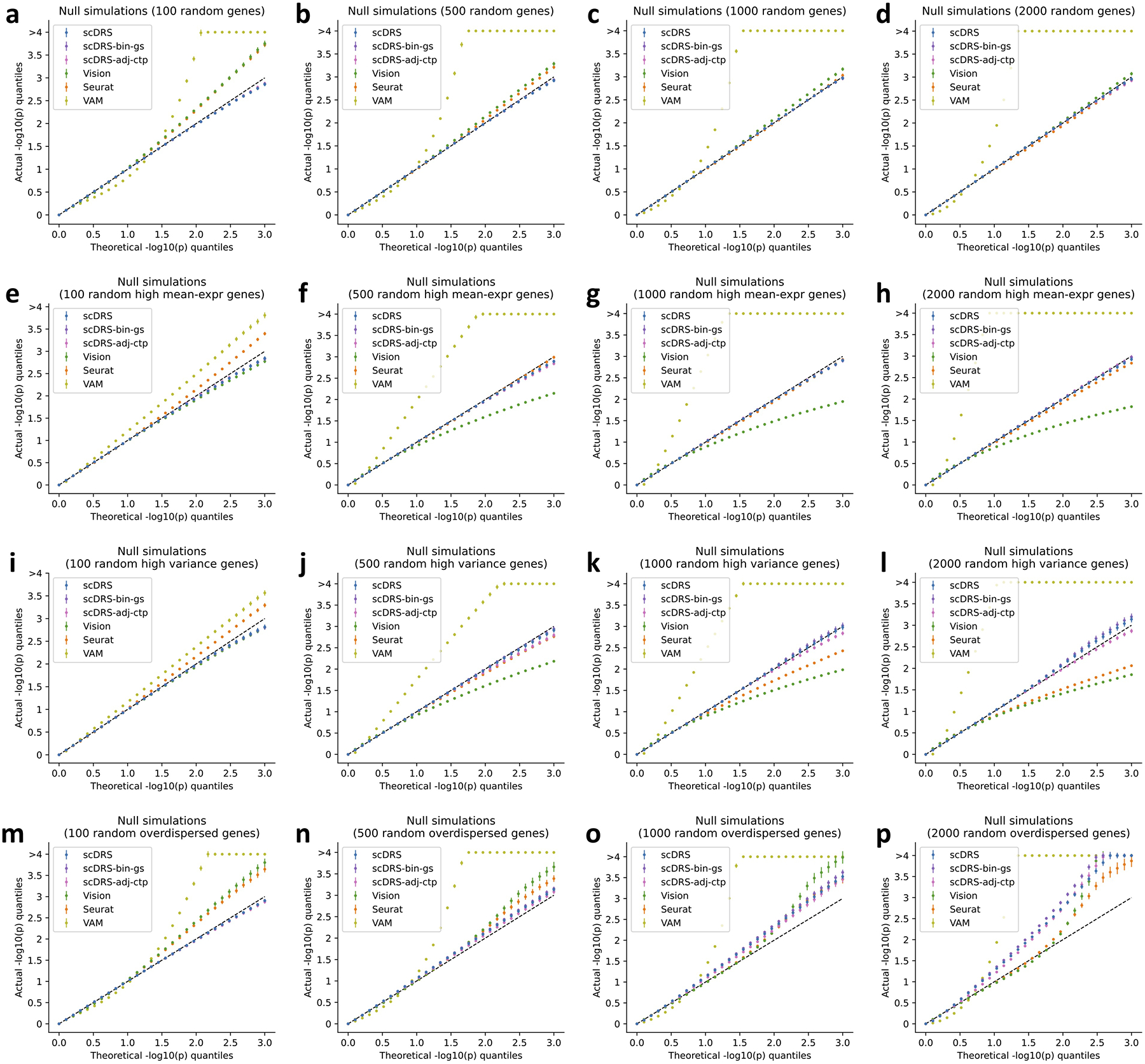
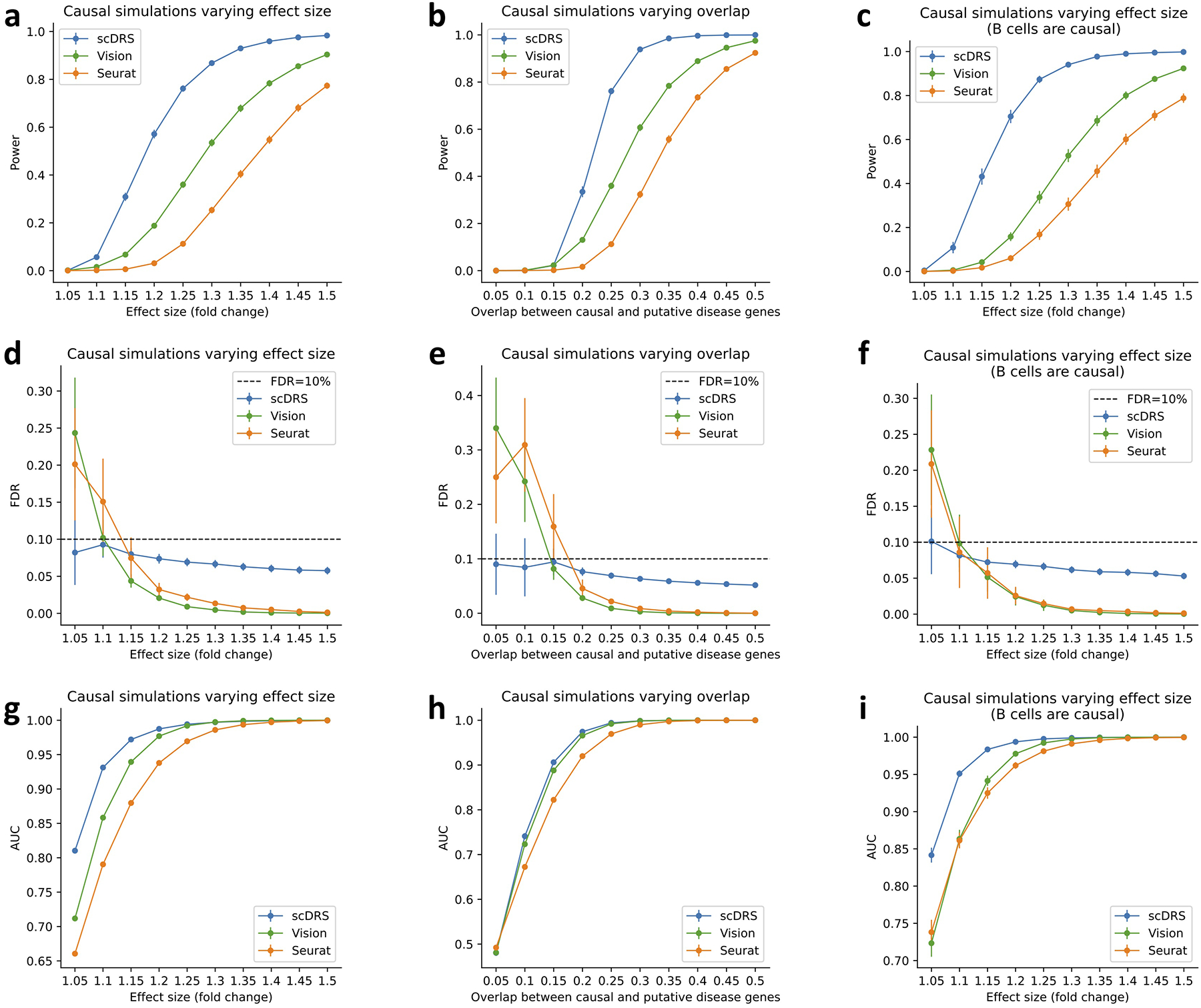
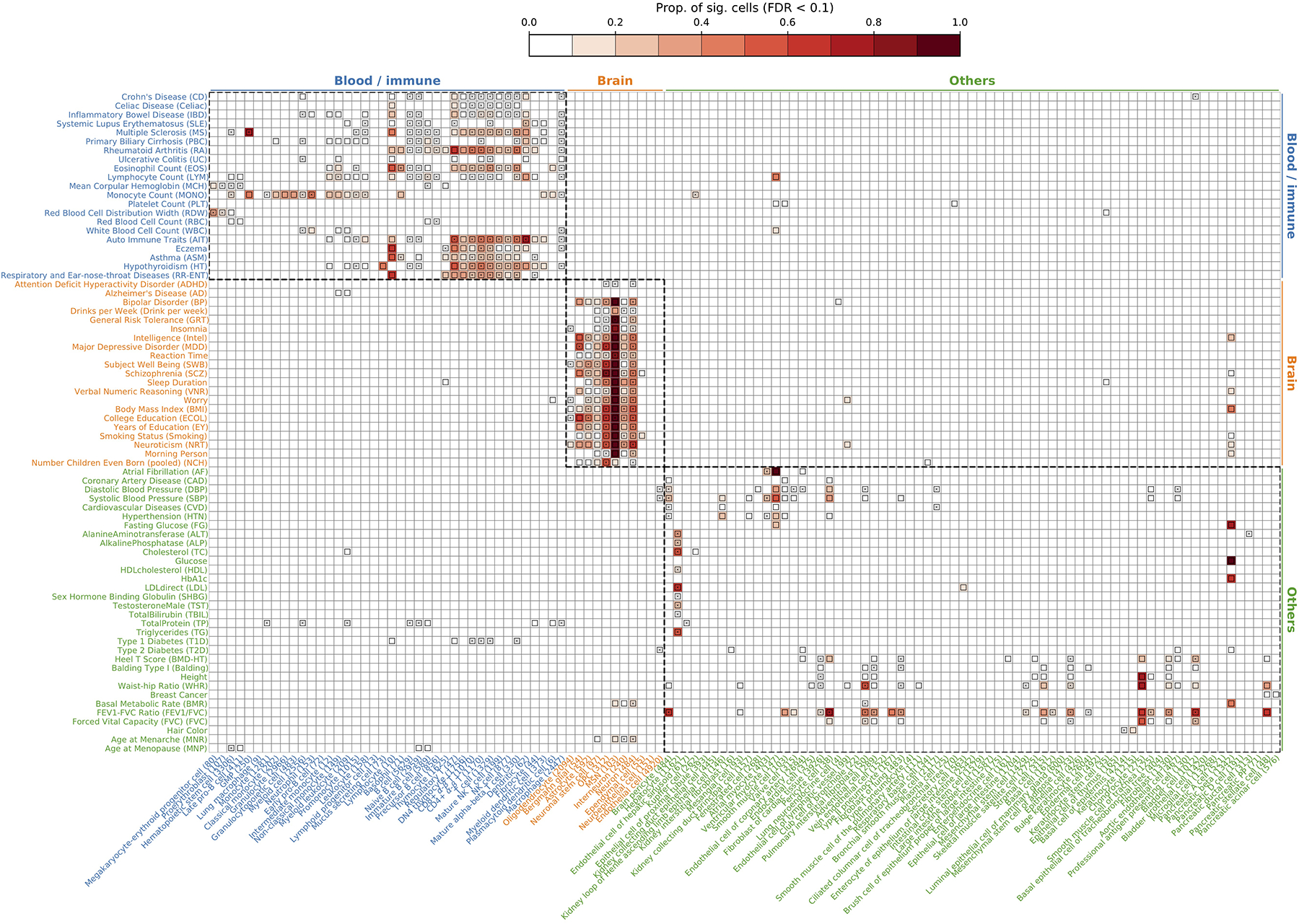
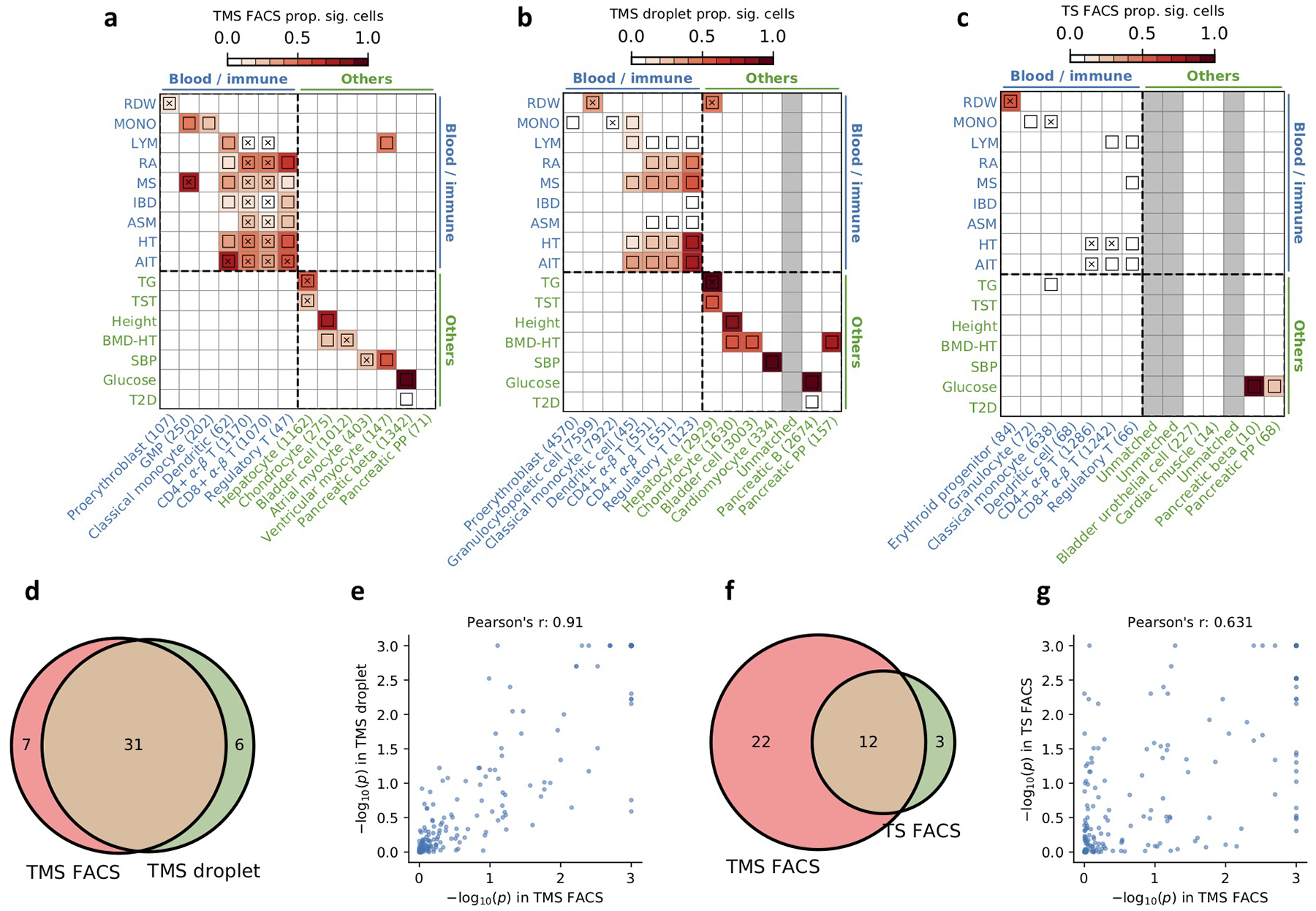
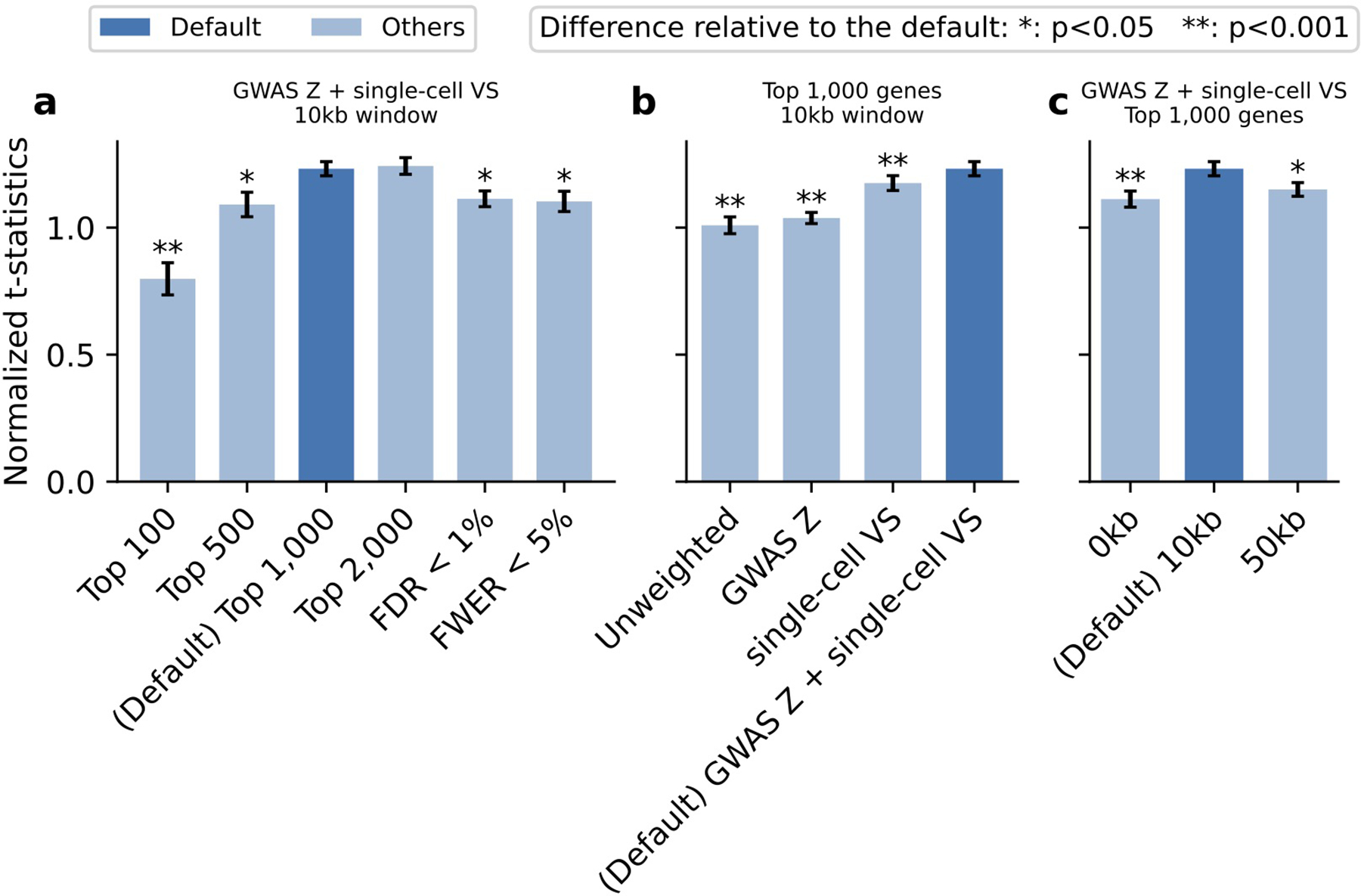
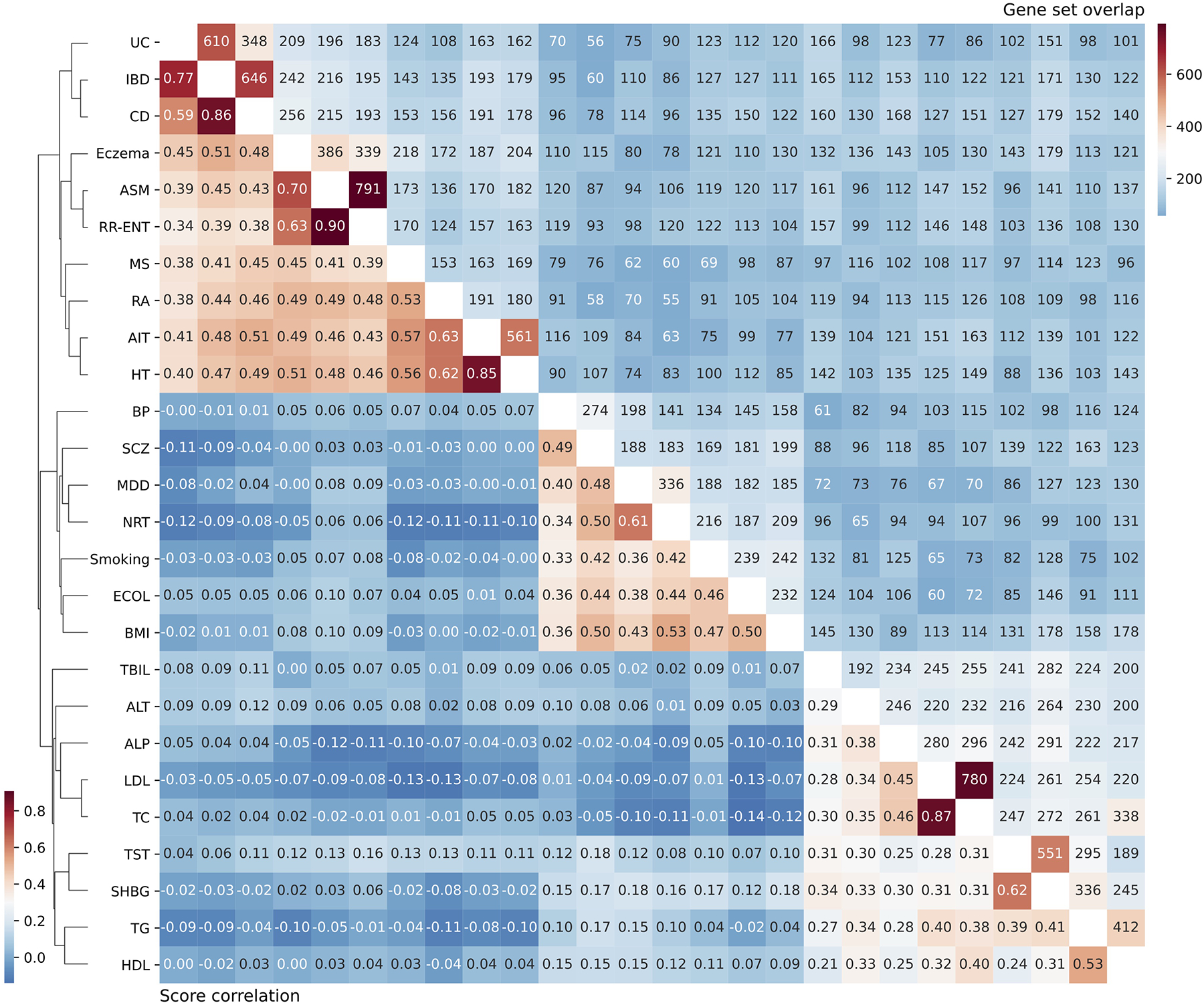
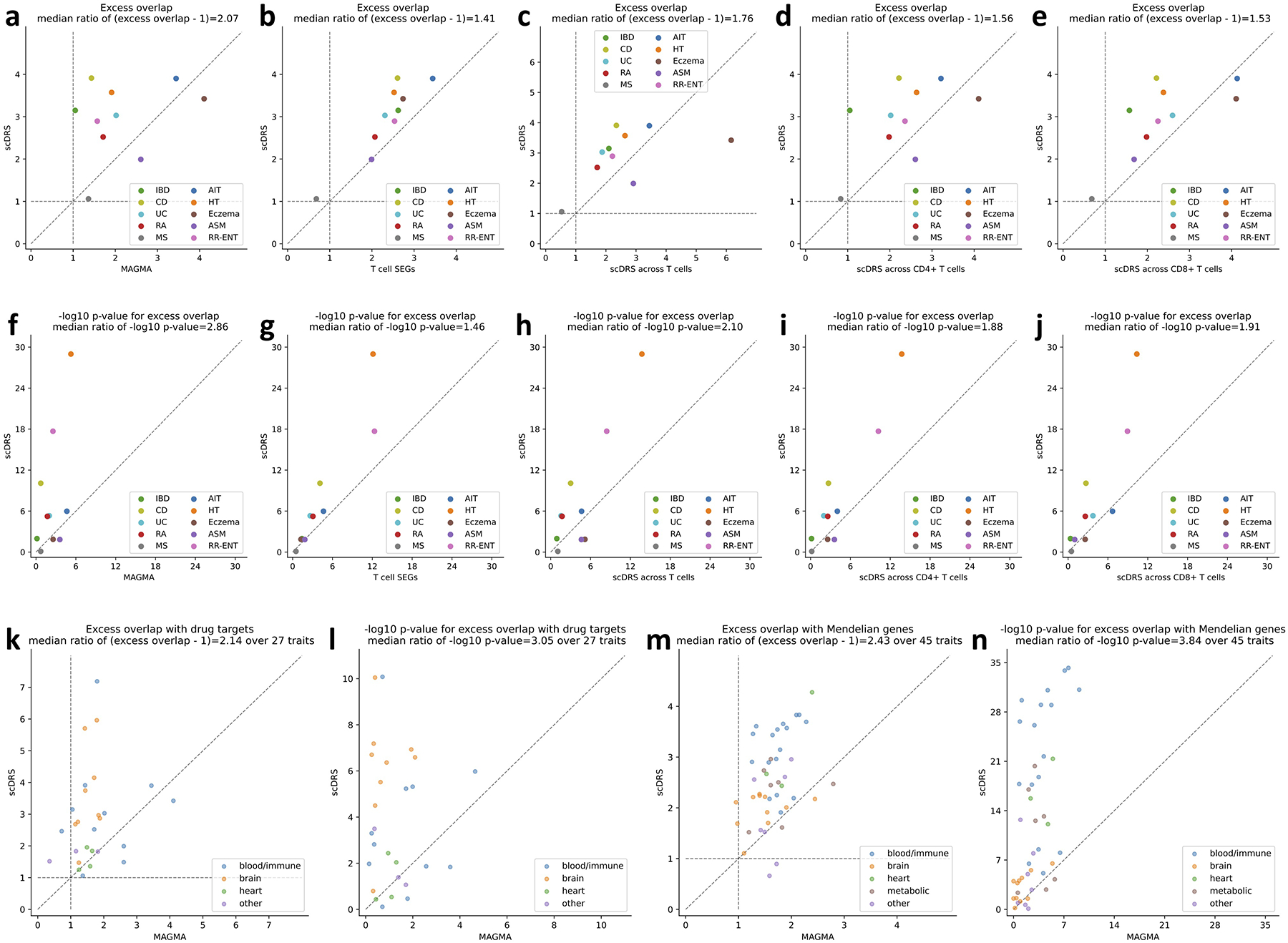
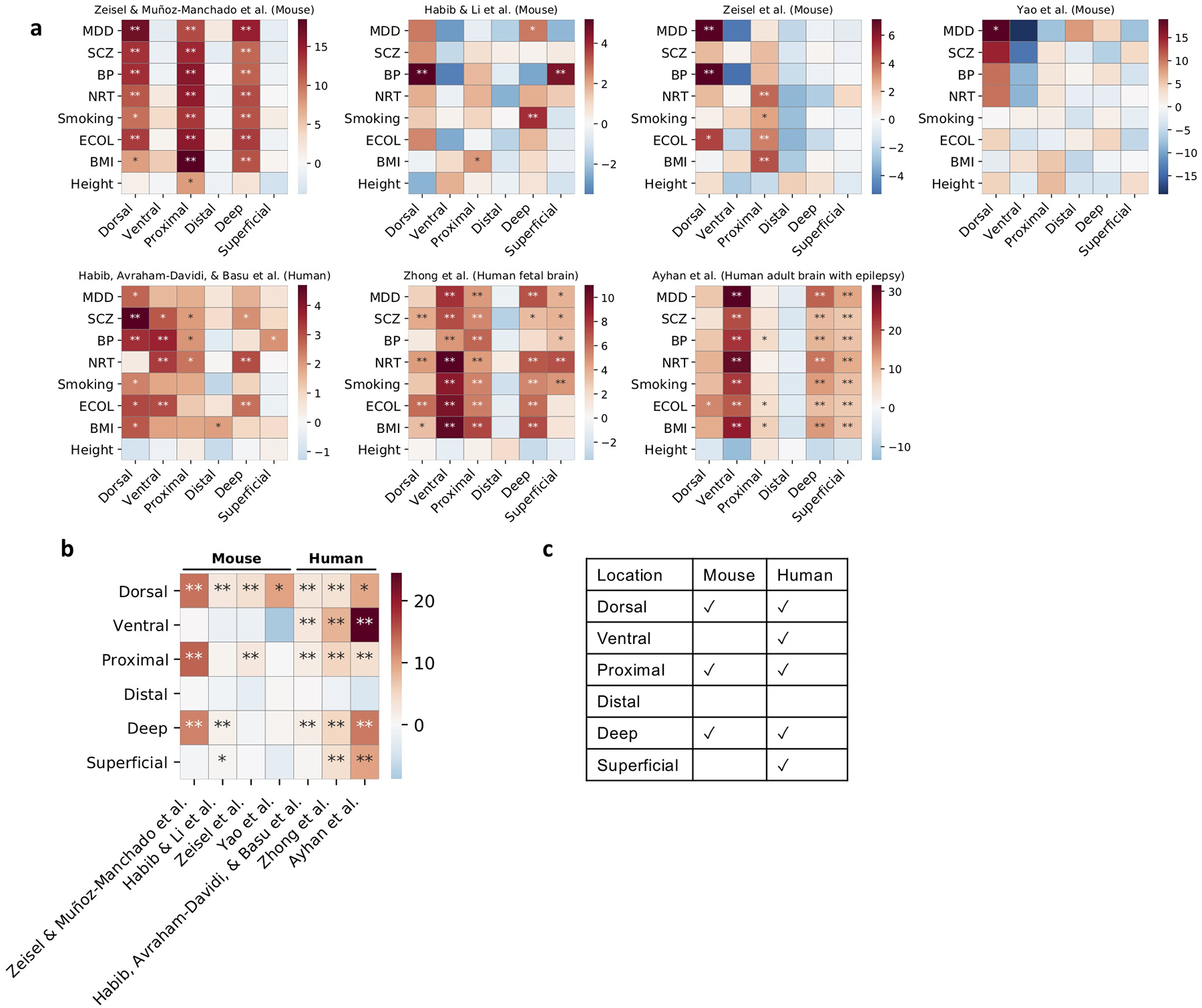
References
-
- Hekselman Idan and Yeger-Lotem Esti. Mechanisms of tissue and cell-type specificity in heritable traits and diseases. Nature Reviews Genetics, 21(3):137–150, 2020. - PubMed
Method-only references
-
- Sakornsakolpat Phuwanat, Prokopenko Dmitry, Lamontagne Maxime, Reeve Nicola F, Guyatt Anna L, Jackson Victoria E, Shrine Nick, Qiao Dandi, Bartz Traci M, Kim Deog Kyeom, et al. Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nature genetics, 51(3):494–505, 2019. - PMC - PubMed
-
- Geary Robert C. The contiguity ratio and statistical mapping. The incorporated statistician, 5(3):115–146, 1954.
-
- Benjamini Yoav and Hochberg Yosef. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal statistical society: series B (Methodological), 57(1):289–300, 1995.
Publication types
MeSH terms
Substances
Grants and funding
- P01 AI148102/AI/NIAID NIH HHS/United States
- F32 HG011434/HG/NHGRI NIH HHS/United States
- R37 MH107649/MH/NIMH NIH HHS/United States
- MC_QA137853/MRC_/Medical Research Council/United Kingdom
- U01 HG012009/HG/NHGRI NIH HHS/United States
- R01 MH121521/MH/NIMH NIH HHS/United States
- T32 HG002295/HG/NHGRI NIH HHS/United States
- R01 MH115676/MH/NIMH NIH HHS/United States
- R01 AR063759/AR/NIAMS NIH HHS/United States
- K99 HG012203/HG/NHGRI NIH HHS/United States
- R01 MH123922/MH/NIMH NIH HHS/United States
- R01 HG006399/HG/NHGRI NIH HHS/United States
- UC2 AR081023/AR/NIAMS NIH HHS/United States
- U01 HG009379/HG/NHGRI NIH HHS/United States
- UH2 AR067677/AR/NIAMS NIH HHS/United States
- MC_PC_17228/MRC_/Medical Research Council/United Kingdom
- R01 MH101244/MH/NIMH NIH HHS/United States
- R01 AG063759/AG/NIA NIH HHS/United States
- T32 HD007470/HD/NICHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
