

Autosomal dominant osteopetrosis type II resulting from a de novo mutation in the CLCN7 gene: A case report
- PMID: 36051116
- PMCID: PMC9297392
- DOI: 10.12998/wjcc.v10.i20.6936
Autosomal dominant osteopetrosis type II resulting from a de novo mutation in the CLCN7 gene: A case report
Abstract
Background: Osteopetrosis is a family of extremely rare diseases caused by failure of osteoclasts and impaired bone resorption. Among them, autosomal dominant osteopetrosis type II (ADO II), related to the chloride channel 7 (CLCN7) gene, is the most frequent form of osteopetrosis. In this study, we report a de novo mutation of CLCN7 in a patient without the family history of ADO II.
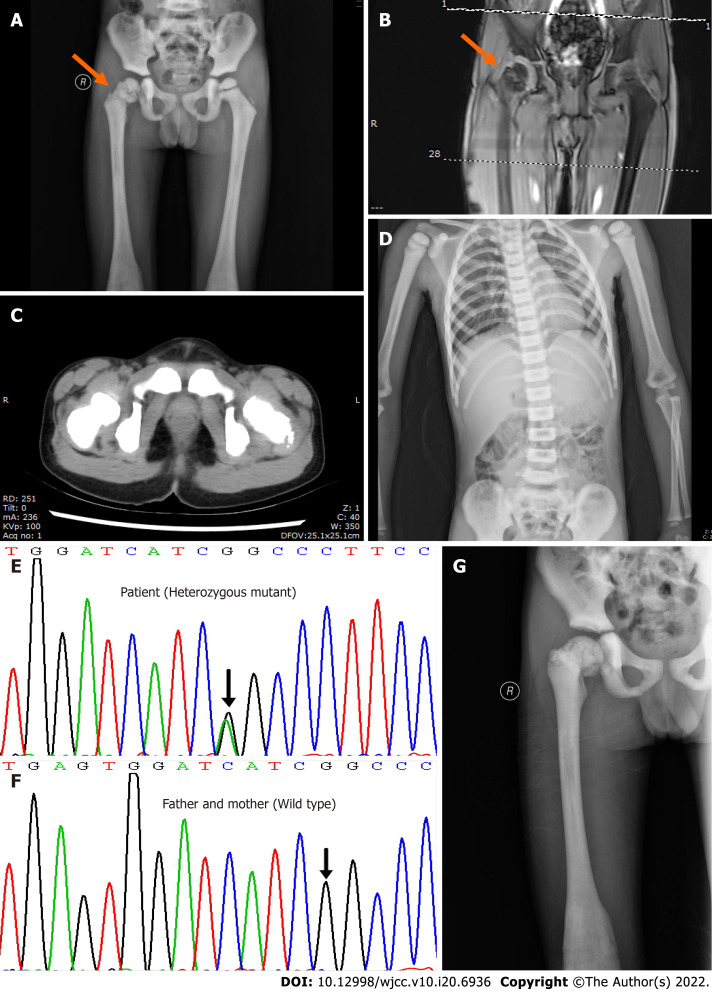
Case summary: A 5-year-old Chinese boy with ADO II was found to have a de novo mutation in the CLCN7 gene [c.746C>T (p.P249L)]. Typical clinical manifestations, including thickening of the cortex of spinal bones and long bones, non-traumatic fracture of the femoral neck, and femoral head necrosis, were found in this patient. The patient is the first reported case of ADO II with the missense mutation c.746C>T (p.P249L) of the CLCN7 gene reported in China. We also review the available literature on ADO II-related CLCN7 mutations, including baseline patient clinical features, special clinical significance, and common mutations.
Conclusion: Our report will enrich the understanding of mutations in ADO II patients. The possibility of a de novo mutation should be considered in individuals who have no family history of osteopetrosis.
Keywords: Autosomal dominant osteopetrosis type Ⅱ; Case report; Chloride channel 7 gene; Osteopetrosis; Whole exome sequencing.
©The Author(s) 2022. Published by Baishideng Publishing Group Inc. All rights reserved.
Conflict of interest statement
Conflict-of-interest statement: All the authors report no relevant conflicts of interest for this article.
Figures
References
-
- Del Fattore A, Cappariello A, Teti A. Genetics, pathogenesis and complications of osteopetrosis. Bone. 2008;42:19–29. - PubMed
-
- Superti-Furga A, Unger S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am J Med Genet A. 2007;143A:1–18. - PubMed
-
- Bollerslev J. Autosomal dominant osteopetrosis: bone metabolism and epidemiological, clinical, and hormonal aspects. Endocr Rev. 1989;10:45–67. - PubMed
-
- Cleiren E, Bénichou O, Van Hul E, Gram J, Bollerslev J, Singer FR, Beaverson K, Aledo A, Whyte MP, Yoneyama T, deVernejoul MC, Van Hul W. Albers-Schönberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the ClCN7 chloride channel gene. Hum Mol Genet. 2001;10:2861–2867. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
