

Transcriptional regulation of Acsl1 by CHREBP and NF-kappa B in macrophages during hyperglycemia and inflammation
- PMID: 36054206
- PMCID: PMC9439225
- DOI: 10.1371/journal.pone.0272986
Transcriptional regulation of Acsl1 by CHREBP and NF-kappa B in macrophages during hyperglycemia and inflammation
Abstract
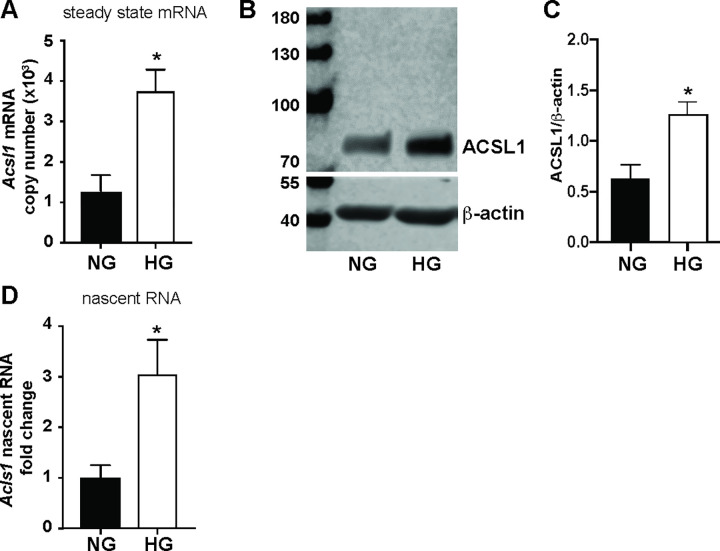
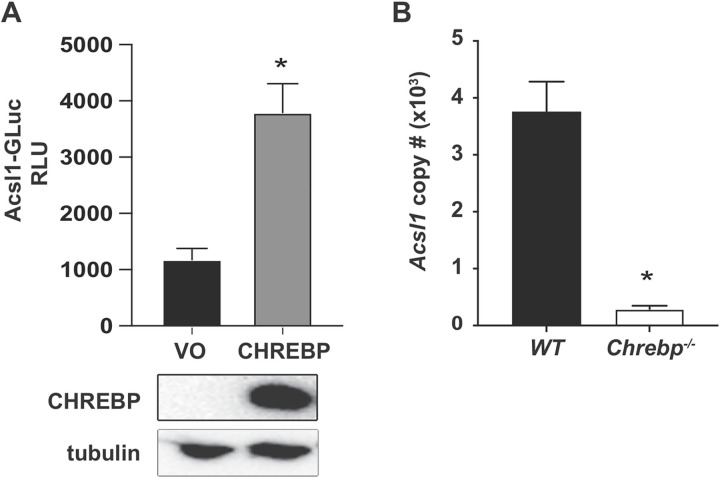
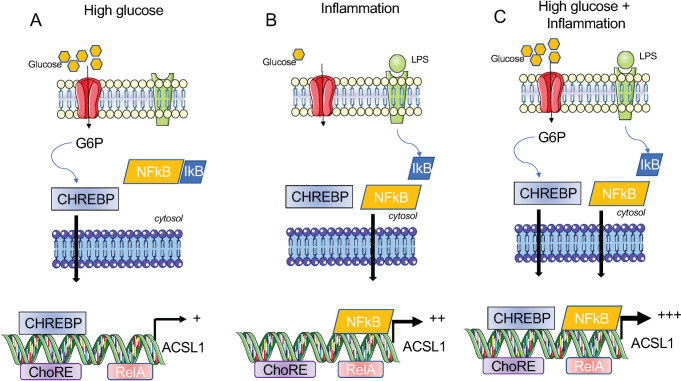
Acyl-CoA synthetase 1 (ACSL1) is an enzyme that converts fatty acids to acyl-CoA-derivatives for lipid catabolism and lipid synthesis in general and can provide substrates for the production of mediators of inflammation in monocytes and macrophages. Acsl1 expression is increased by hyperglycemia and inflammatory stimuli in monocytes and macrophages, and promotes the pro-atherosclerotic effects of diabetes in mice. Yet, surprisingly little is known about the mechanisms underlying Acsl1 transcriptional regulation. Here we demonstrate that the glucose-sensing transcription factor, Carbohydrate Response Element Binding Protein (CHREBP), is a regulator of the expression of Acsl1 mRNA by high glucose in mouse bone marrow-derived macrophages (BMDMs). In addition, we show that inflammatory stimulation of BMDMs with lipopolysaccharide (LPS) increases Acsl1 mRNA via the transcription factor, NF-kappa B. LPS treatment also increases ACSL1 protein abundance and localization to membranes where it can exert its activity. Using an Acsl1 reporter gene containing the promoter and an upstream regulatory region, which has multiple conserved CHREBP and NF-kappa B (p65/RELA) binding sites, we found increased Acsl1 promoter activity upon CHREBP and p65/RELA expression. We also show that CHREBP and p65/RELA occupy the Acsl1 promoter in BMDMs. In primary human monocytes cultured in high glucose versus normal glucose, ACSL1 mRNA expression was elevated by high glucose and further enhanced by LPS treatment. Our findings demonstrate that CHREBP and NF-kappa B control Acsl1 expression under hyperglycemic and inflammatory conditions.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures




References
-
- Kanter JE, Kramer F, Barnhart S, Averill MM, Vivekanandan-Giri A, Vickery T, et al.. Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc Natl Acad Sci U S A. 2012;109(12):E715–24. Epub 2012/02/07. doi: 10.1073/pnas.1111600109 ; PubMed Central PMCID: PMC3311324. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
