

Molecular dynamics simulations highlight the altered binding landscape at the spike-ACE2 interface between the Delta and Omicron variants compared to the SARS-CoV-2 original strain
- PMID: 36055162
- PMCID: PMC9420038
- DOI: 10.1016/j.compbiomed.2022.106035
Molecular dynamics simulations highlight the altered binding landscape at the spike-ACE2 interface between the Delta and Omicron variants compared to the SARS-CoV-2 original strain
Abstract
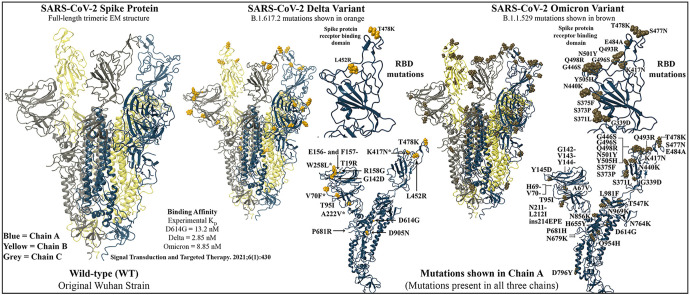
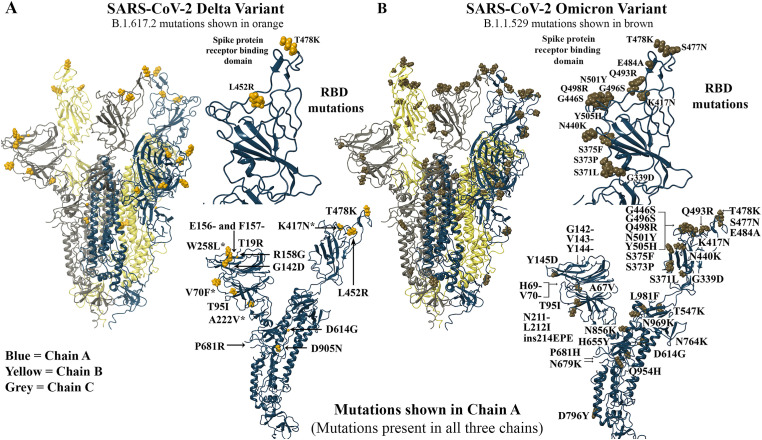
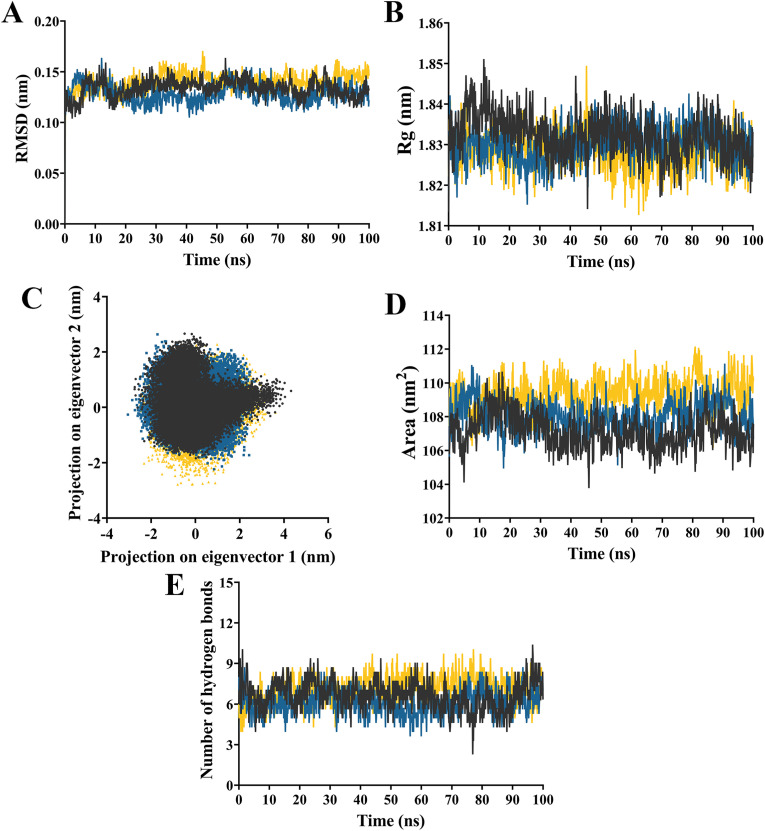
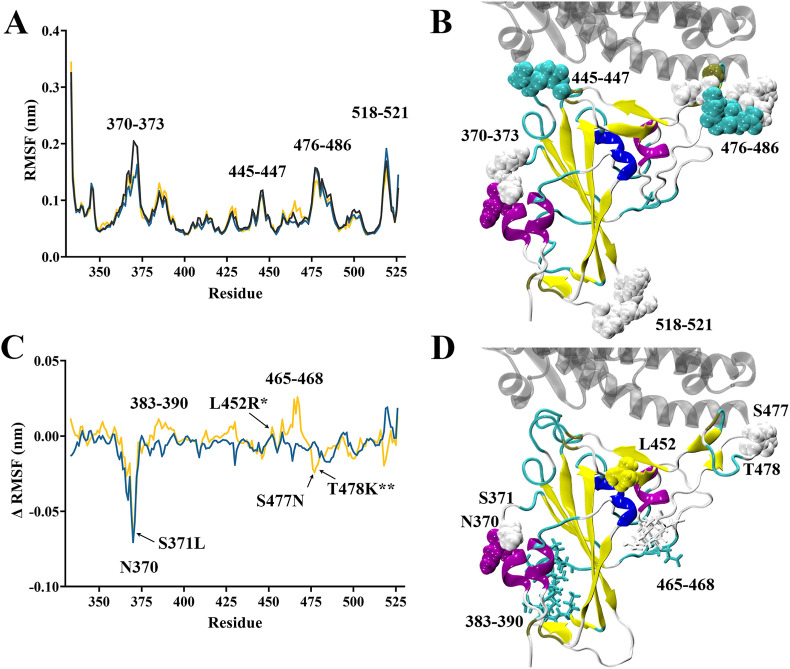
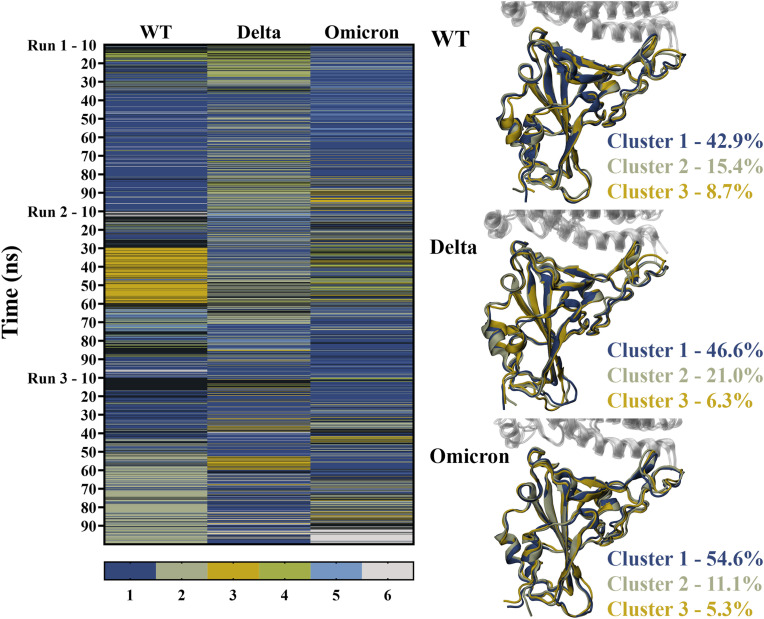
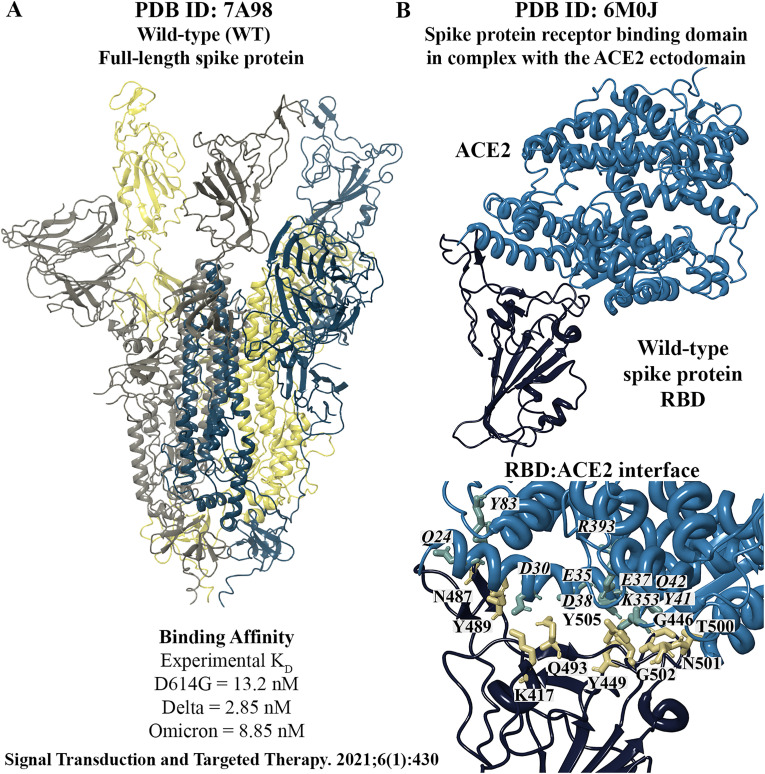
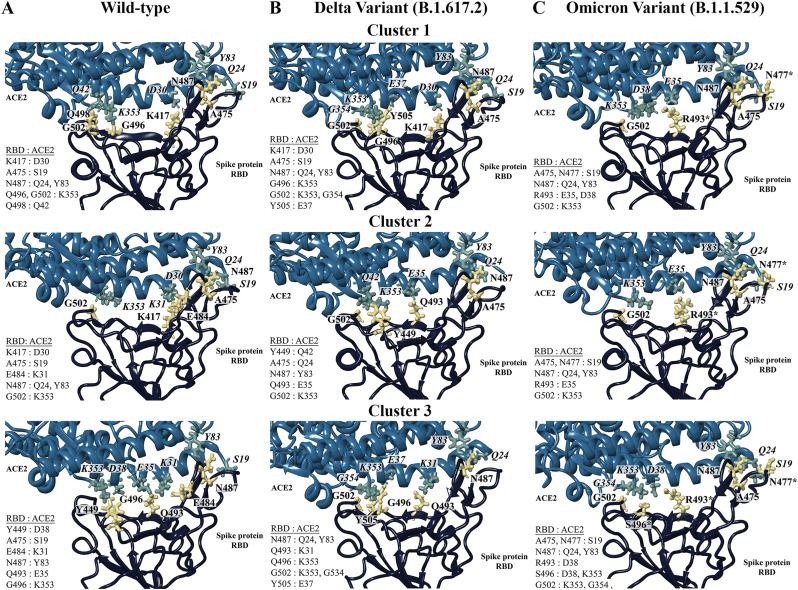
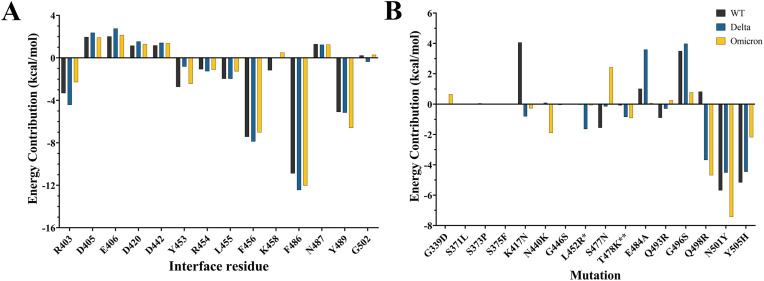
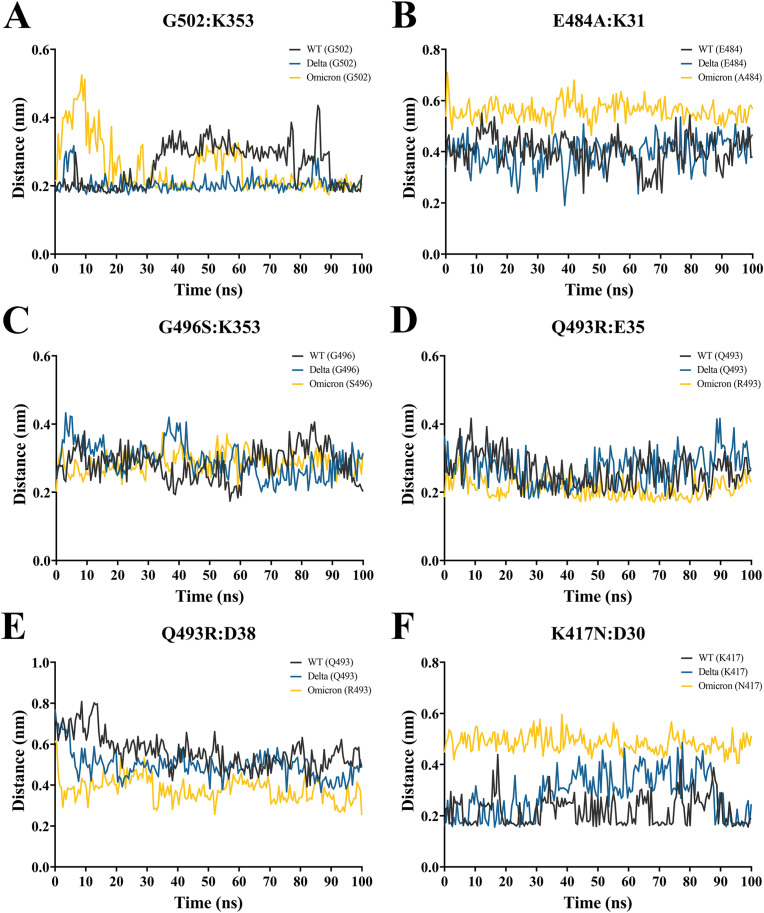
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) B.1.1.529 variant (Omicron), represents a significant deviation in genetic makeup and function compared to previous variants. Following the BA.1 sublineage, the BA.2 and BA.3 Omicron subvariants became dominant, and currently the BA.4 and BA.5, which are quite distinct variants, have emerged. Using molecular dynamics simulations, we investigated the binding characteristics of the Delta and Omicron (BA.1) variants in comparison to wild-type (WT) at the interface of the spike protein receptor binding domain (RBD) and human angiotensin converting enzyme-2 (ACE2) ectodomain. The primary aim was to compare our molecular modelling systems with previously published observations, to determine the robustness of our approach for rapid prediction of emerging future variants. Delta and Omicron were found to bind to ACE2 with similar affinities (-39.4 and -43.3 kcal/mol, respectively) and stronger than WT (-33.5 kcal/mol). In line with previously published observations, the energy contributions of the non-mutated residues at the interface were largely retained between WT and the variants, with F456, F486, and Y489 having the strongest energy contributions to ACE2 binding. Further, residues N440K, Q498R, and N501Y were predicted to be energetically favourable in Omicron. In contrast to Omicron, which had the E484A and K417N mutations, intermolecular bonds were detected for the residue pairs E484:K31 and K417:D30 in WT and Delta, in accordance with previously published findings. Overall, our simplified molecular modelling approach represents a step towards predictive model systems for rapidly analysing arising variants of concern.
Keywords: ACE2 receptors; Delta variant; Omicron variant; SARS-CoV-2; Spike protein.
Copyright © 2022 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of competing interest There are no conflicts to declare.
Figures
Similar articles
-
Exploring conformational landscapes and binding mechanisms of convergent evolution for the SARS-CoV-2 spike Omicron variant complexes with the ACE2 receptor using AlphaFold2-based structural ensembles and molecular dynamics simulations.Phys Chem Chem Phys. 2024 Jun 26;26(25):17720-17744. doi: 10.1039/d4cp01372g. Phys Chem Chem Phys. 2024. PMID: 38869513
-
AlphaFold2 Modeling and Molecular Dynamics Simulations of the Conformational Ensembles for the SARS-CoV-2 Spike Omicron JN.1, KP.2 and KP.3 Variants: Mutational Profiling of Binding Energetics Reveals Epistatic Drivers of the ACE2 Affinity and Escape Hotspots of Antibody Resistance.Viruses. 2024 Sep 13;16(9):1458. doi: 10.3390/v16091458. Viruses. 2024. PMID: 39339934 Free PMC article.
-
Omicron and Delta variant of SARS-CoV-2: A comparative computational study of spike protein.J Med Virol. 2022 Apr;94(4):1641-1649. doi: 10.1002/jmv.27526. Epub 2021 Dec 27. J Med Virol. 2022. PMID: 34914115
-
The Biological Functions and Clinical Significance of SARS-CoV-2 Variants of Corcern.Front Med (Lausanne). 2022 May 20;9:849217. doi: 10.3389/fmed.2022.849217. eCollection 2022. Front Med (Lausanne). 2022. PMID: 35669924 Free PMC article. Review.
-
Distinct Conformations of SARS-CoV-2 Omicron Spike Protein and Its Interaction with ACE2 and Antibody.Int J Mol Sci. 2023 Feb 14;24(4):3774. doi: 10.3390/ijms24043774. Int J Mol Sci. 2023. PMID: 36835186 Free PMC article. Review.
Cited by
-
Identification of medicinal plant-based phytochemicals as a potential inhibitor for SARS-CoV-2 main protease (Mpro) using molecular docking and deep learning methods.Comput Biol Med. 2023 May;157:106785. doi: 10.1016/j.compbiomed.2023.106785. Epub 2023 Mar 11. Comput Biol Med. 2023. PMID: 36931201 Free PMC article.
-
Different aspects in explaining how mutations could affect the binding mechanism of receptor binding domain of SARS-CoV-2 spike protein in interaction with ACE2.PLoS One. 2023 Sep 8;18(9):e0291210. doi: 10.1371/journal.pone.0291210. eCollection 2023. PLoS One. 2023. PMID: 37682927 Free PMC article.
-
Cooperative and structural relationships of the trimeric Spike with infectivity and antibody escape of the strains Delta (B.1.617.2) and Omicron (BA.2, BA.5, and BQ.1).J Comput Aided Mol Des. 2023 Dec;37(12):585-606. doi: 10.1007/s10822-023-00534-0. Epub 2023 Oct 4. J Comput Aided Mol Des. 2023. PMID: 37792106
-
Dramatic Differences between the Structural Susceptibility of the S1 Pre- and S2 Postfusion States of the SARS-CoV-2 Spike Protein to External Electric Fields Revealed by Molecular Dynamics Simulations.Viruses. 2023 Dec 11;15(12):2405. doi: 10.3390/v15122405. Viruses. 2023. PMID: 38140646 Free PMC article.
-
Adding a Twist to Lateral Flow Immunoassays: A Direct Replacement of Antibodies with Helical Affibodies, from Selection to Application.J Am Chem Soc. 2025 Apr 9;147(14):11925-11940. doi: 10.1021/jacs.4c17452. Epub 2025 Mar 26. J Am Chem Soc. 2025. PMID: 40135773 Free PMC article.
References
-
- Tracking W.H.O. World Health Organization; 2021. SARS-CoV-2 Variants.
-
- Centers for Disease Control and Prevention. SARS-CoV-2 variant classifications and definitions 2021 [updated Dec 1 2021]. Available from: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classificatio....
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
