

State-of-the-art therapies for Rett syndrome
- PMID: 36056801
- PMCID: PMC10087176
- DOI: 10.1111/dmcn.15383
State-of-the-art therapies for Rett syndrome
Abstract
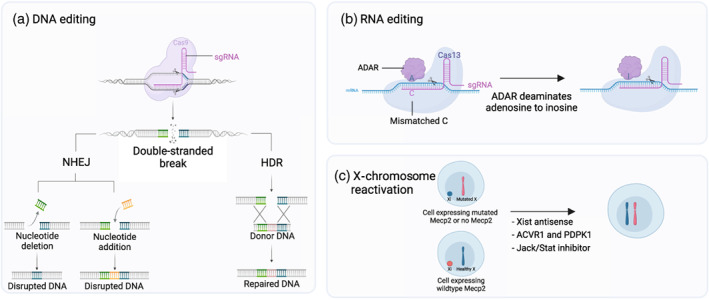
Rett syndrome (RTT) is an X-linked neurogenetic disorder caused by mutations of the MECP2 (methyl-CpG-binding protein 2) gene. Over two decades of work established MeCP2 as a protein with pivotal roles in the regulation of the epigenome, neuronal physiology, synaptic maintenance, and behaviour. Given the genetic aetiology of RTT and the proof of concept of its reversal in a mouse model, considerable efforts have been made to design therapeutic approaches to re-express MeCP2. By being at the forefront of the development of innovative gene therapies, research on RTT is of paramount importance for the treatment of monogenic neurological diseases. Here we discuss the recent advances and challenges of promising genetic strategies for the treatment of RTT including gene replacement therapies, gene/RNA editing strategies, and reactivation of the silenced X chromosome. WHAT THIS PAPER ADDS: Recent advances shed light on the promises of gene replacement therapy with new vectors designed to control the levels of MeCP2 expression. New developments in DNA/RNA editing approaches or reactivation of the silenced X chromosome open the possibility to re-express the native MeCP2 locus at endogenous levels. Current strategies still face limitations in transduction efficiency and future work is needed to improve brain delivery.
© 2022 The Authors. Developmental Medicine & Child Neurology published by John Wiley & Sons Ltd on behalf of Mac Keith Press.
Figures
References
-
- Rett A. [On a unusual brain atrophy syndrome in hyperammonemia in childhood]. Wiener medizinische Wochenschrift 1966. ; 116: 723–726 - PubMed
-
- Hagberg B, Aicardi J, Dias K and Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Annals of Neurology 1983; 14: 471–479. - PubMed
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U and Zoghbi HY. Rett syndrome is caused by mutations in X‐linked MECP2, encoding methyl‐CpG‐binding protein 2. Nature Genetics 1999; 23: 185–188. - PubMed
-
- Lyst MJ, Bird A. Rett syndrome: a complex disorder with simple roots. Nat Rev Genet 2015; 16: 261‐75. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
