

Taz protects hematopoietic stem cells from an aging-dependent decrease in PU.1 activity
- PMID: 36057685
- PMCID: PMC9440927
- DOI: 10.1038/s41467-022-32970-1
Taz protects hematopoietic stem cells from an aging-dependent decrease in PU.1 activity
Abstract
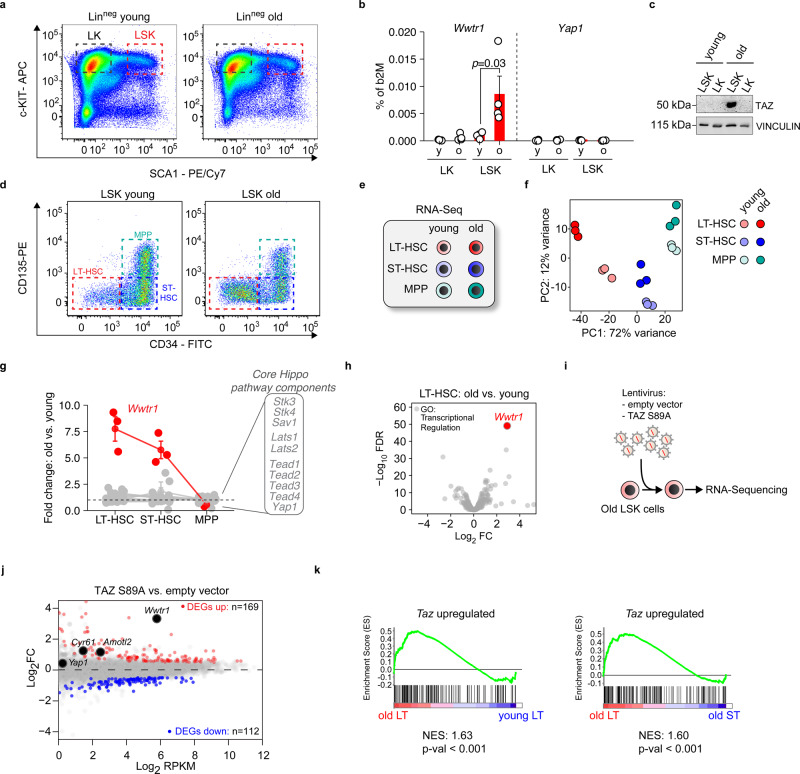
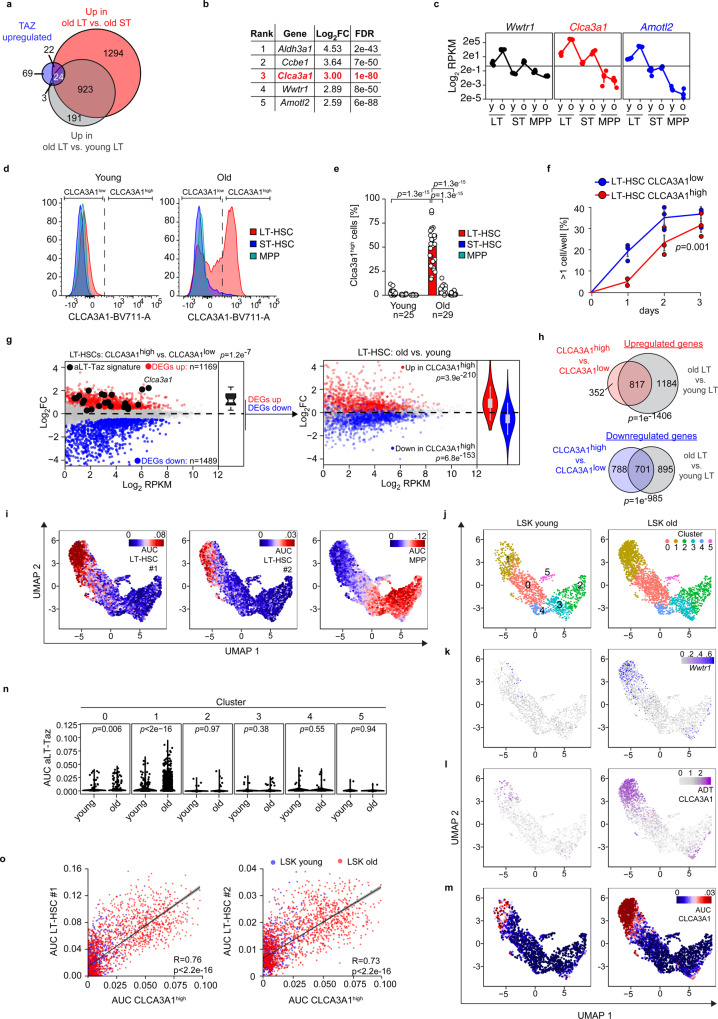
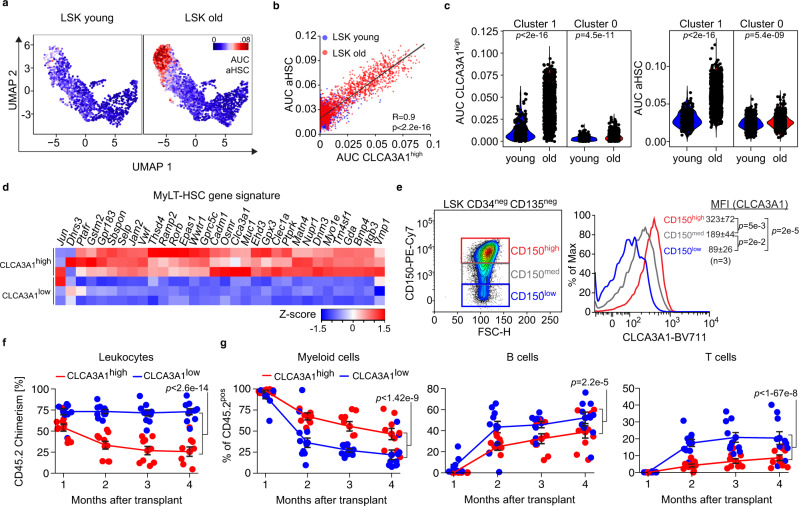
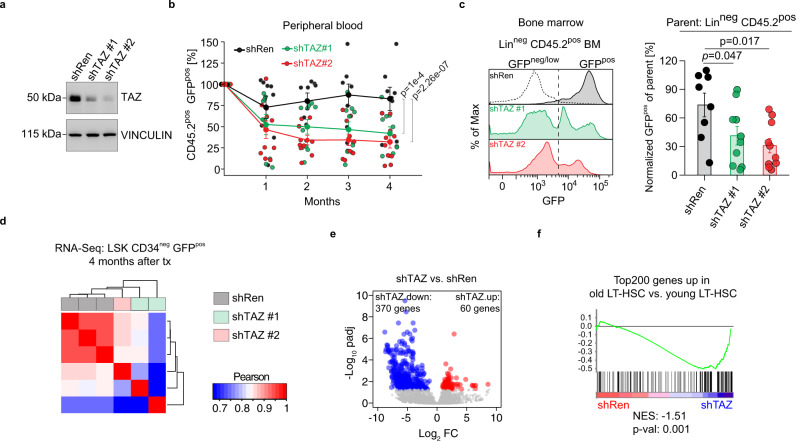
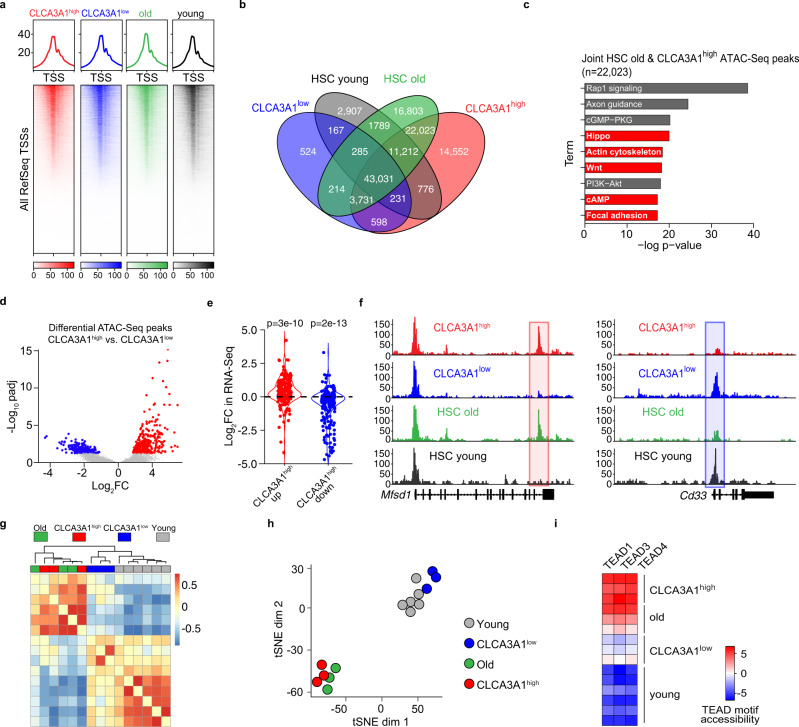
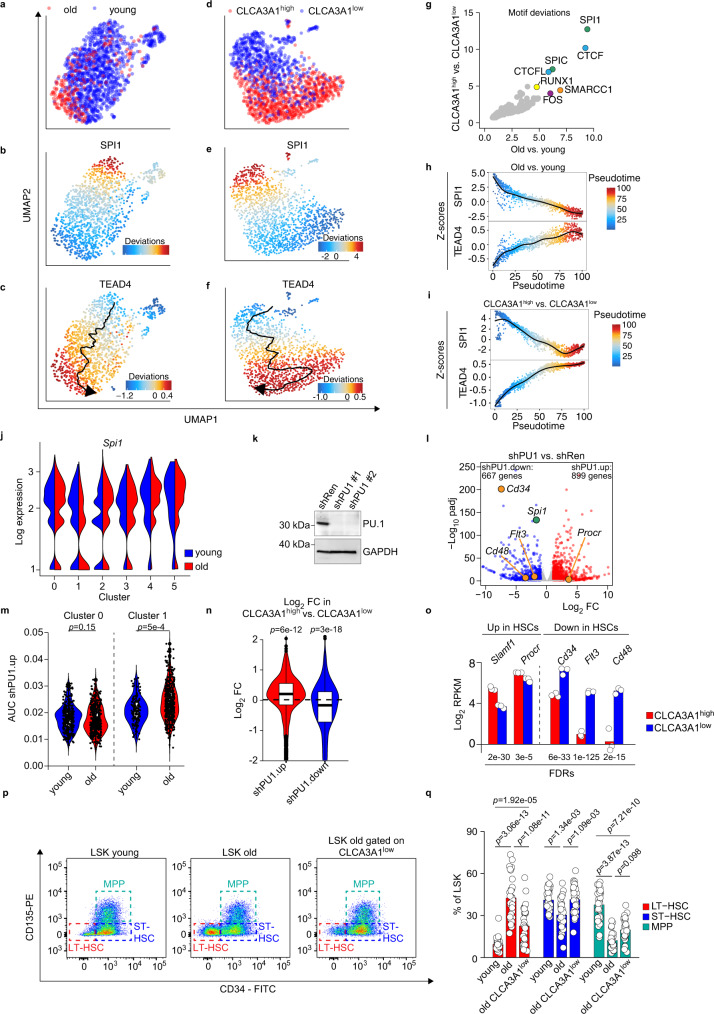
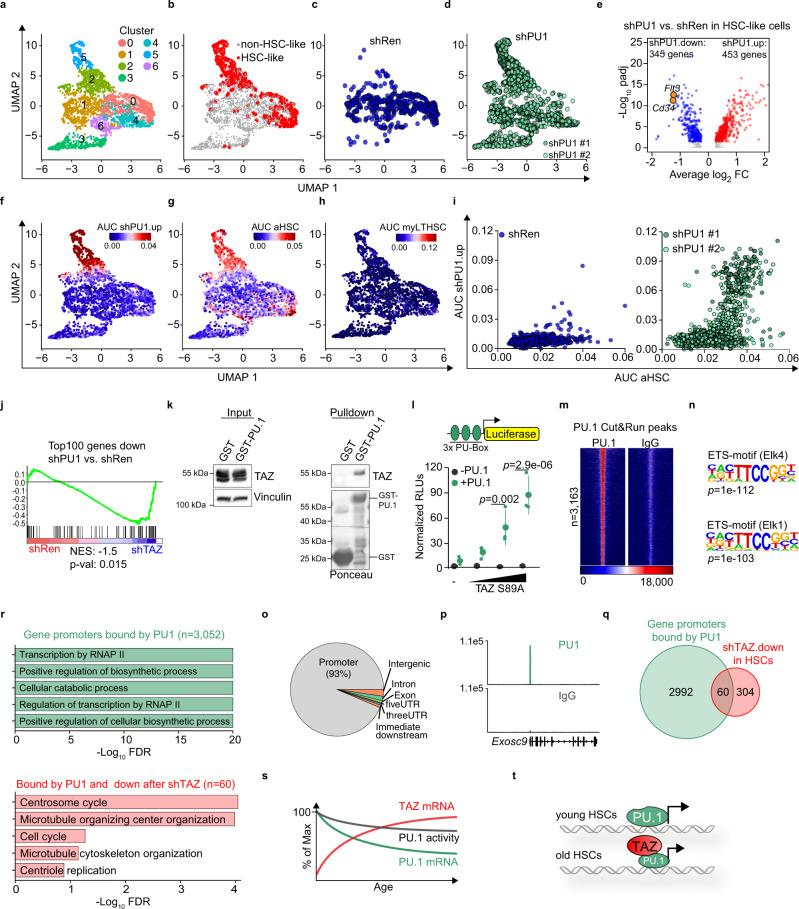
Specific functions of the immune system are essential to protect us from infections caused by pathogens such as viruses and bacteria. However, as we age, the immune system shows a functional decline that can be attributed in large part to age-associated defects in hematopoietic stem cells (HSCs)-the cells at the apex of the immune cell hierarchy. Here, we find that the Hippo pathway coactivator TAZ is potently induced in old HSCs and protects these cells from functional decline. We identify Clca3a1 as a TAZ-induced gene that allows us to trace TAZ activity in vivo. Using CLCA3A1 as a marker, we can isolate "young-like" HSCs from old mice. Mechanistically, Taz acts as coactivator of PU.1 and to some extent counteracts the gradual loss of PU.1 expression during HSC aging. Our work thus uncovers an essential role for Taz in a previously undescribed fail-safe mechanism in aging HSCs.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
