

Predicting accurate ab initio DNA electron densities with equivariant neural networks
- PMID: 36057785
- PMCID: PMC9674991
- DOI: 10.1016/j.bpj.2022.08.045
Predicting accurate ab initio DNA electron densities with equivariant neural networks
Abstract
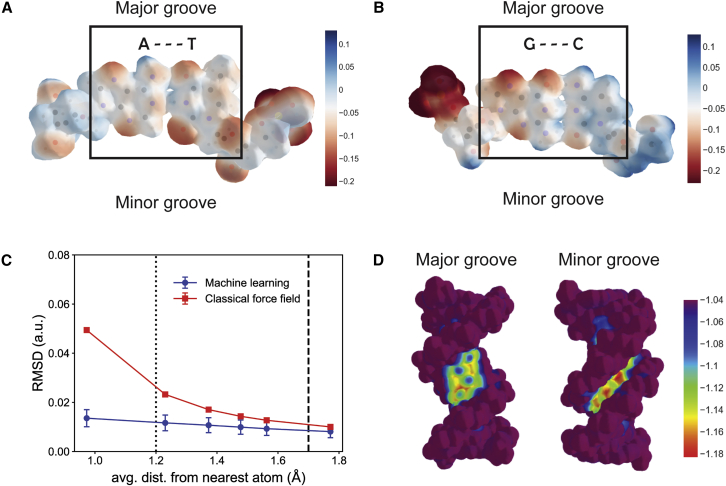
One of the fundamental limitations of accurately modeling biomolecules like DNA is the inability to perform quantum chemistry calculations on large molecular structures. We present a machine learning model based on an equivariant Euclidean neural network framework to obtain accurate ab initio electron densities for arbitrary DNA structures that are much too large for conventional quantum methods. The model is trained on representative B-DNA basepair steps that capture both base pairing and base stacking interactions. The model produces accurate electron densities for arbitrary B-DNA structures with typical errors of less than 1%. Crucially, the error does not increase with system size, which suggests that the model can extrapolate to large DNA structures with negligible loss of accuracy. The model also generalizes reasonably to other DNA structural motifs such as the A- and Z-DNA forms, despite being trained on only B-DNA configurations. The model is used to calculate electron densities of several large-scale DNA structures, and we show that the computational scaling for this model is essentially linear. We also show that this machine learning electron density model can be used to calculate accurate electrostatic potentials for DNA. These electrostatic potentials produce more accurate results compared with classical force fields and do not show the usual deficiencies at short range.
Copyright © 2022 Biophysical Society. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
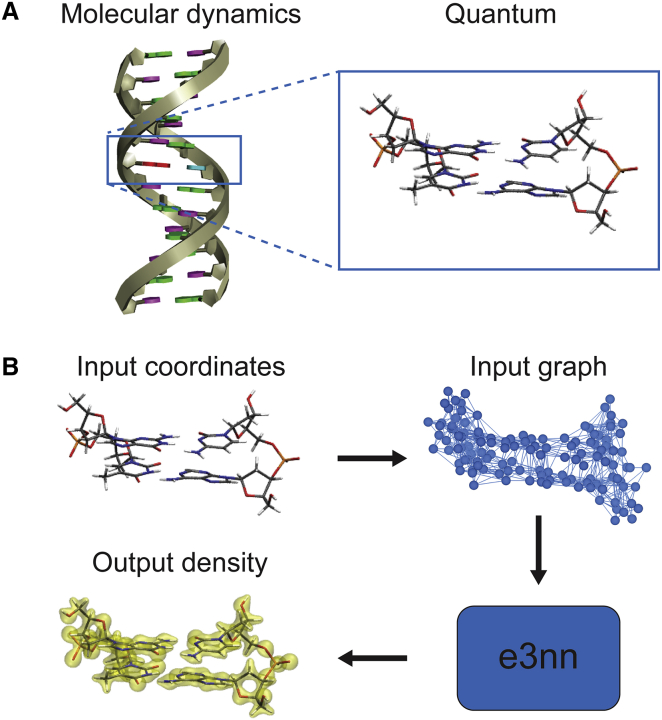
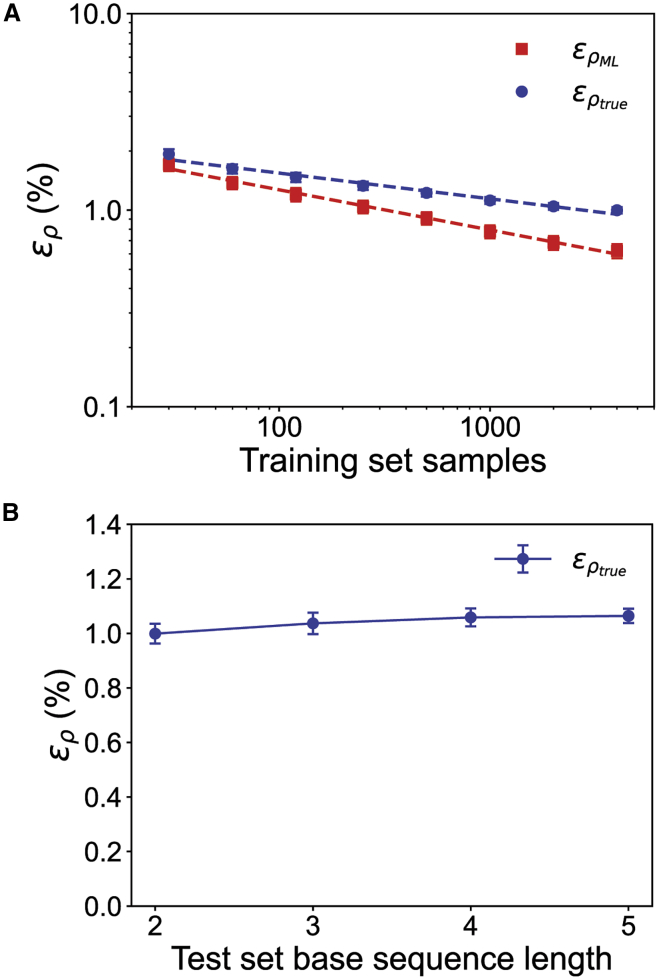
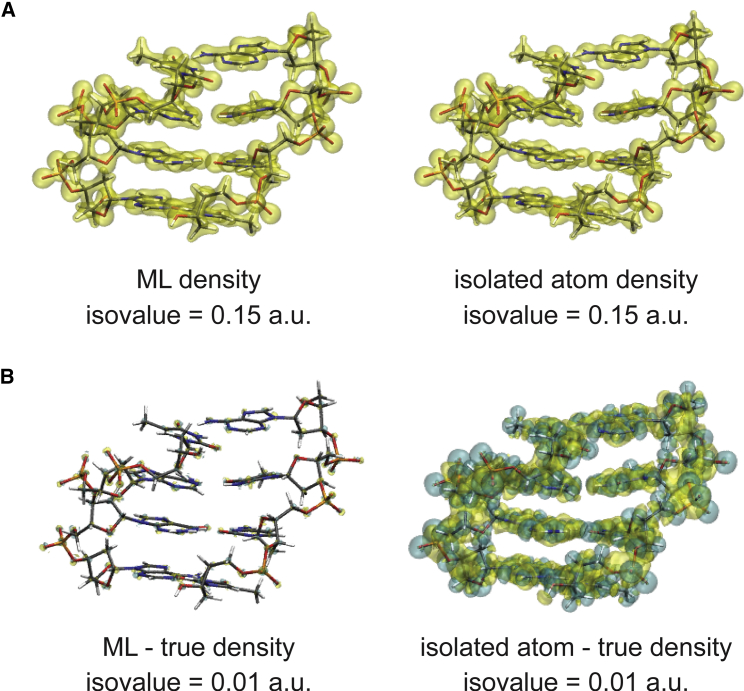
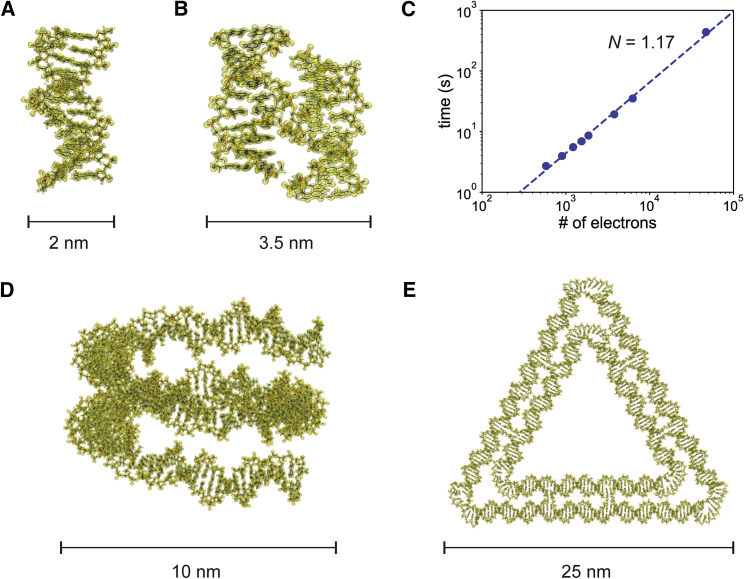
References
-
- Cole D.J., Hine N.D.M. Applications of large-scale density functional theory in biology. J. Phys. Condens. Matter. 2016;28:393001. - PubMed
-
- González J., Baños I., et al. Millán J. Unravelling protein–DNA interactions at molecular level: a DFT and NCI study. J. Chem. Theor. Comput. 2016;12:523–534. - PubMed
-
- Liu X.W., Li J., Ji L.N., et al. Experimental and theoretical study on DNA-binding and photocleavage properties of chiral complexes Δ- and Λ-(Ru(bpy)2L) (L = o-hpip, m-hpip and p-hpip) Dalton Trans. 2003:1352–1359.
-
- Kruse H., Banáš P., Šponer J. Investigations of stacked DNA base-pair steps: highly accurate stacking interaction energies, energy decomposition, and many-body stacking effects. J. Chem. Theor. Comput. 2019;15:95–115. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
