

Interleukin-18 signaling promotes activation of hepatic stellate cells in mouse liver fibrosis
- PMID: 36059147
- PMCID: PMC9984672
- DOI: 10.1002/hep.32776
Interleukin-18 signaling promotes activation of hepatic stellate cells in mouse liver fibrosis
Abstract
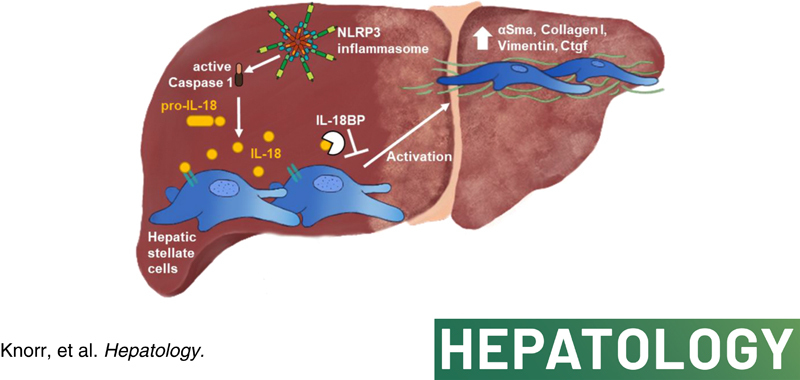
Background and aims: Nucleotide-binding oligomerization domain-like receptor-family pyrin domain-containing 3 (NLRP3) inflammasome activation has been shown to result in liver fibrosis. Mechanisms and downstream signaling remain incompletely understood. Here, we studied the role of IL-18 in hepatic stellate cells (HSCs), and its impact on liver fibrosis.
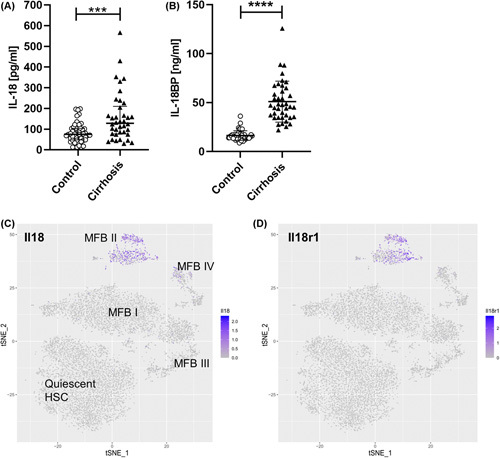
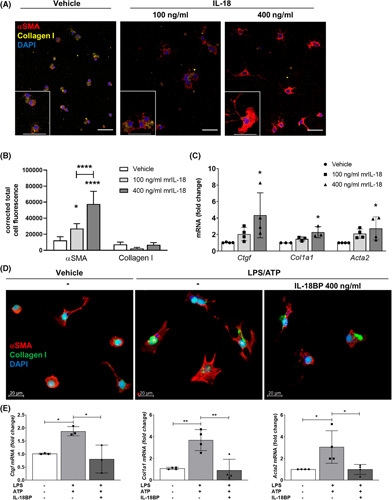
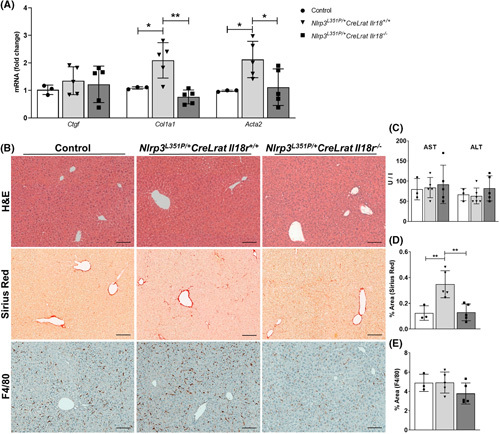
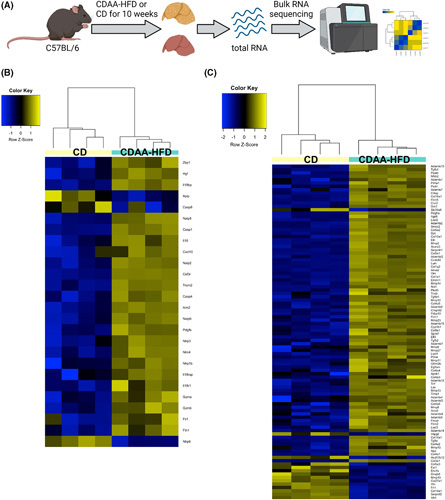
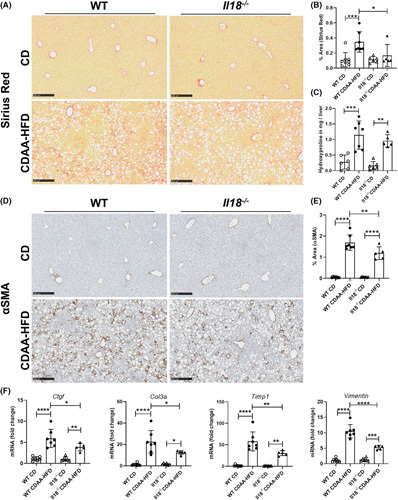
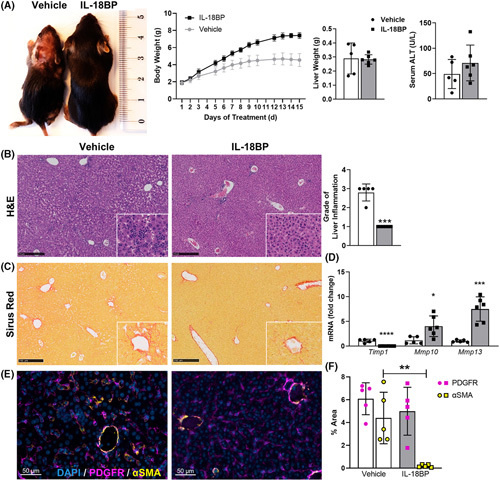
Approach and results: We observed significantly increased serum levels of IL-18 (128.4 pg/ml vs. 74.9 pg/ml) and IL-18 binding protein (BP; 46.50 ng/ml vs. 15.35 ng/ml) in patients with liver cirrhosis compared with healthy controls. Single cell RNA sequencing data showed that an immunoregulatory subset of murine HSCs highly expresses Il18 and Il18r1 . Treatment of cultured primary murine HSC with recombinant mouse IL-18 accelerated their transdifferentiation into myofibroblasts. In vivo , IL-18 receptor-deficient mice had reduced liver fibrosis in a model of fibrosis induced by HSC-specific NLRP3 overactivation. Whole liver RNA sequencing analysis from a murine model of severe NASH-induced fibrosis by feeding a choline-deficient, L-amino acid-defined, high fat diet showed that genes related to IL-18 and its downstream signaling were significantly upregulated, and Il18-/- mice receiving this diet for 10 weeks showed protection from fibrotic changes with decreased number of alpha smooth muscle actin-positive cells and collagen deposition. HSC activation triggered by NLRP3 inflammasome activation was abrogated when IL-18 signaling was blocked by its naturally occurring antagonist IL-18BP. Accordingly, we observed that the severe inflammatory phenotype associated with myeloid cell-specific NLRP3 gain-of-function was rescued by IL-18BP.
Conclusions: Our study highlights the role of IL-18 in the development of liver fibrosis by its direct effect on HSC activation identifying IL-18 as a target to treat liver fibrosis.
Copyright © 2023 The Author(s). Published by Wolters Kluwer Health, Inc.
Conflict of interest statement
Frank Tacke advises and received grants from Gilead. He consults for Novo Nordisk and Pfizer. He is on the speakers’ bureau for Falk and AbbVie. He received grants from Inventiva, Bristol‐Myers Squibb, and Allergan. Hal M. Hoffman consults for Novartis.
Figures
References
-
- Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, et al. Cloning of a new cytokine that induces IFN‐gamma production by T cells. Nature. 1995;378(6552):88–91. - PubMed
-
- Ghayur T, Banerjee S, Hugunin M, Butler D, Herzog L, Carter A, et al. Caspase‐1 processes IFN‐gamma‐inducing factor and regulates LPS‐induced IFN‐gamma production. Nature. 1997;386(6625):619–23. - PubMed
-
- Mehta VB, Hart J, Wewers MD. ATP‐stimulated release of interleukin (IL)‐1beta and IL‐18 requires priming by lipopolysaccharide and is independent of caspase‐1 cleavage. J Biol Chem. 2001;276(6):3820–6. - PubMed
