

Inferring Phenotypic Trait Evolution on Large Trees With Many Incomplete Measurements
- PMID: 36060555
- PMCID: PMC9438787
- DOI: 10.1080/01621459.2020.1799812
Inferring Phenotypic Trait Evolution on Large Trees With Many Incomplete Measurements
Abstract
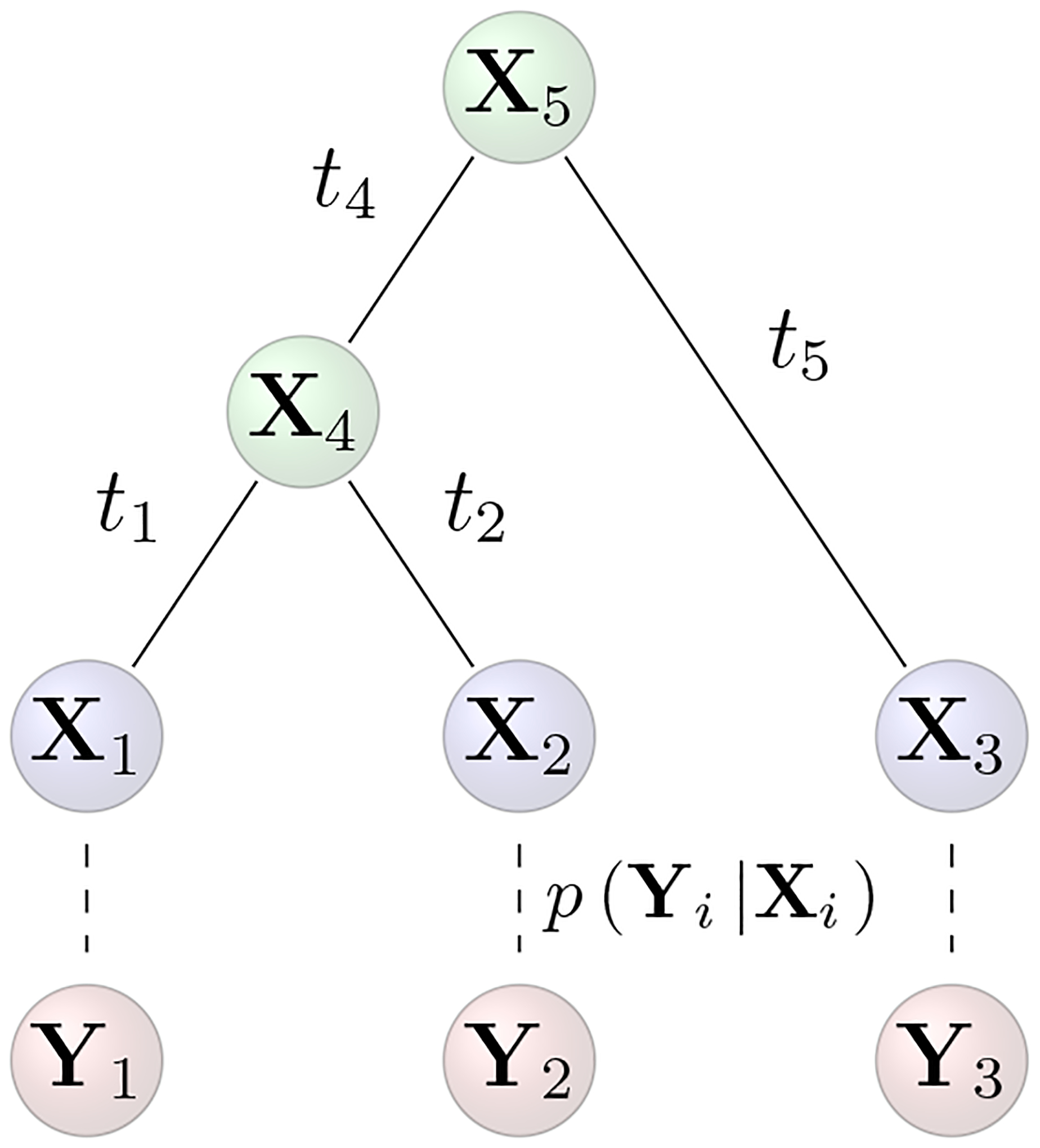
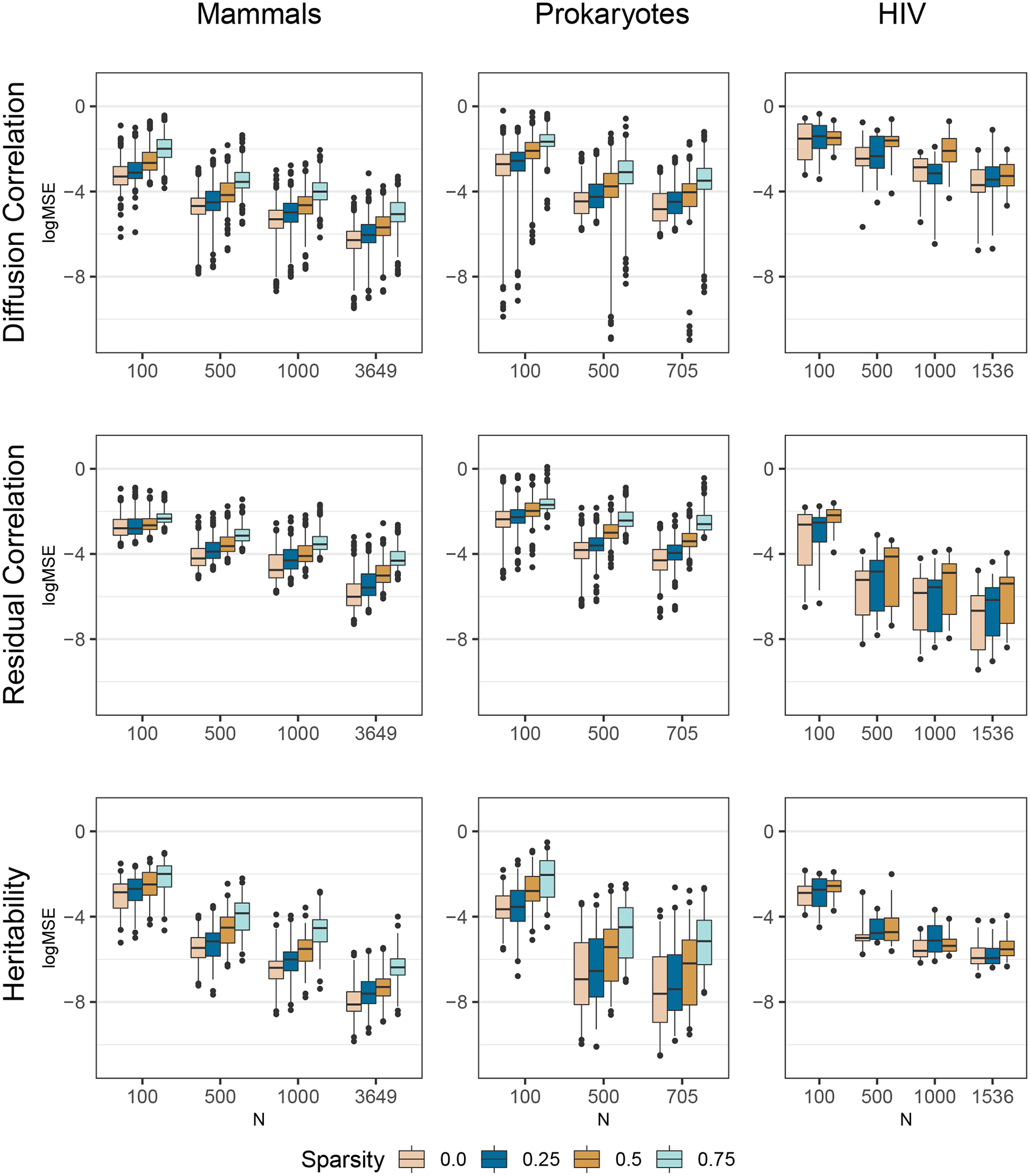
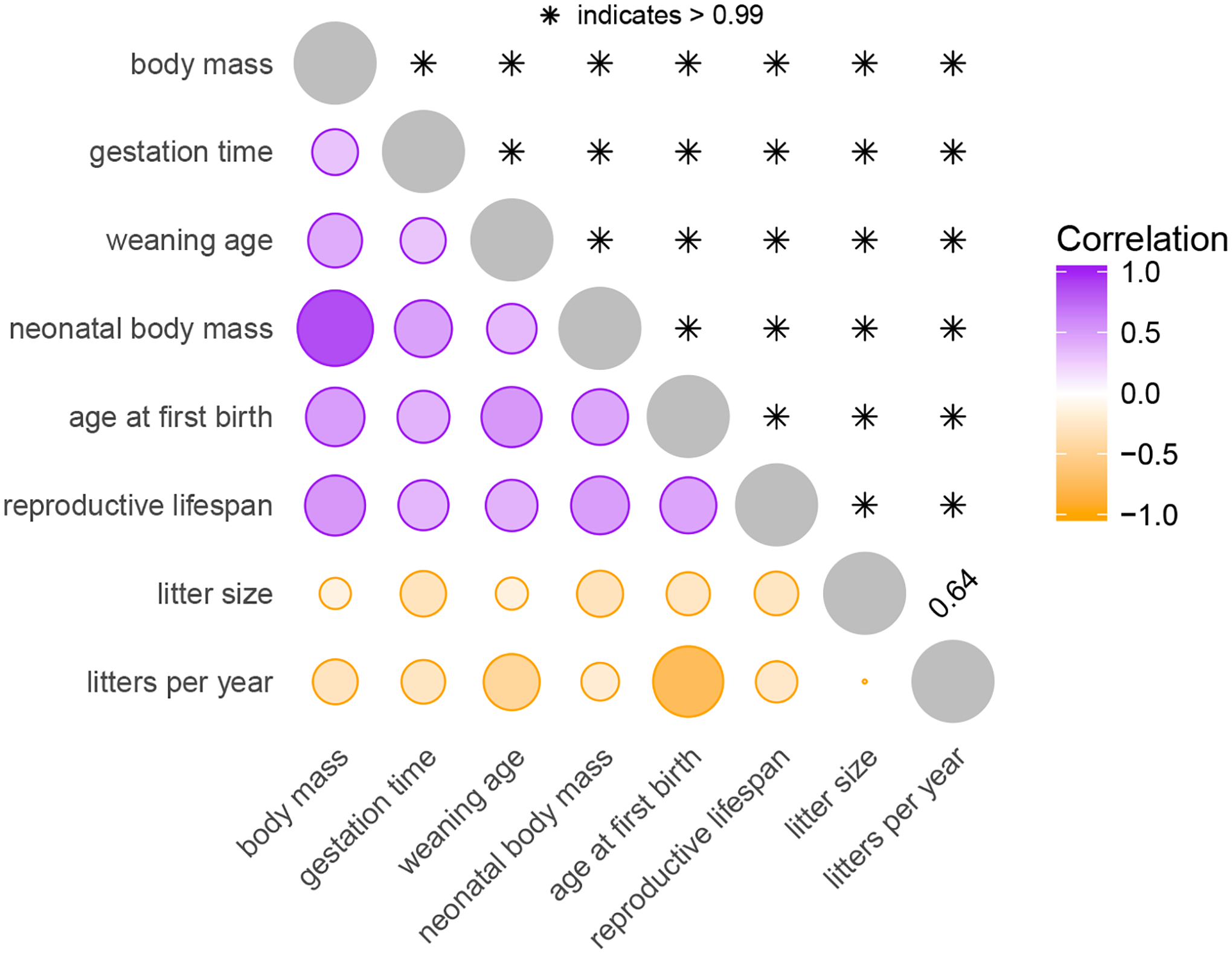
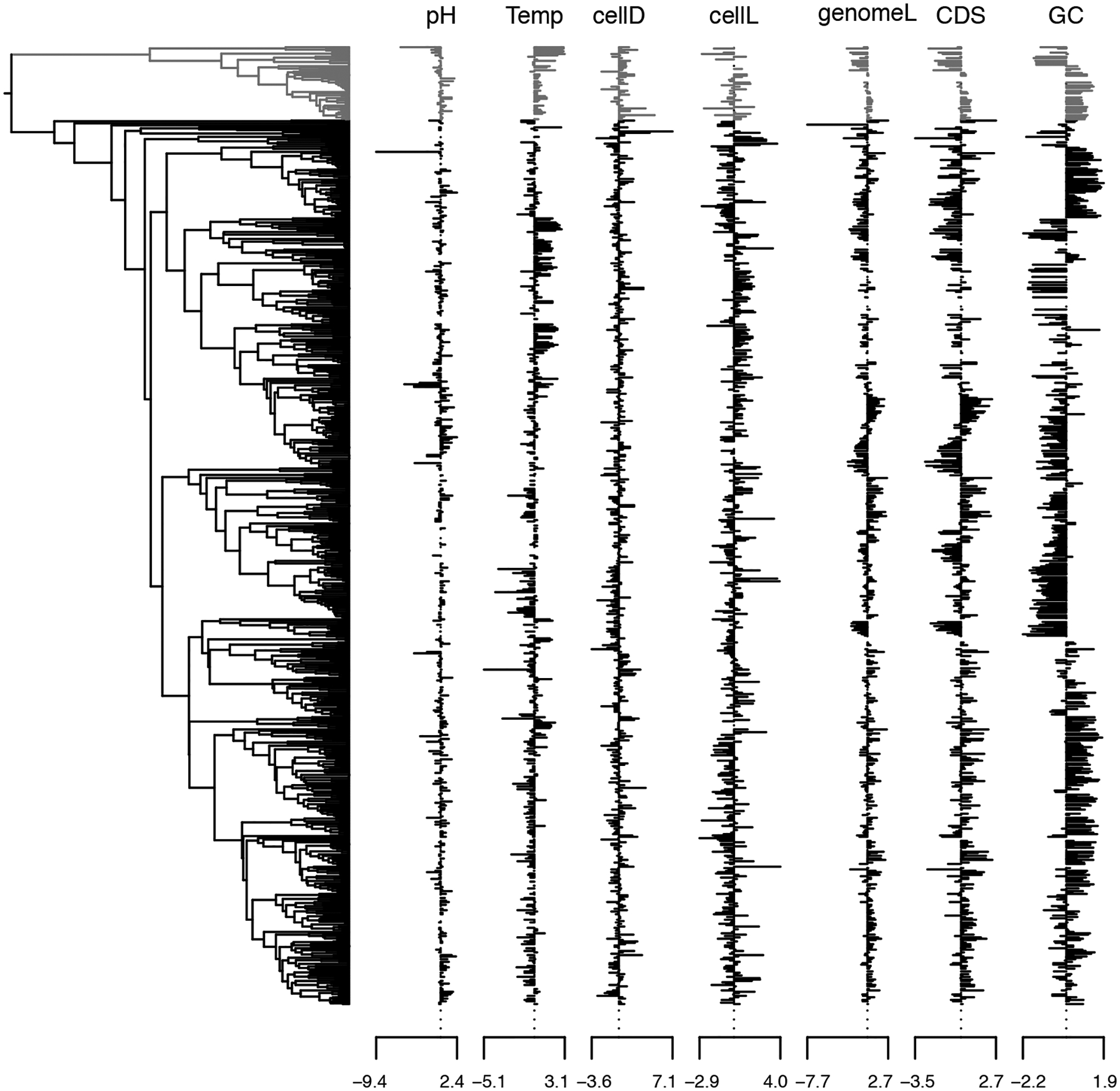
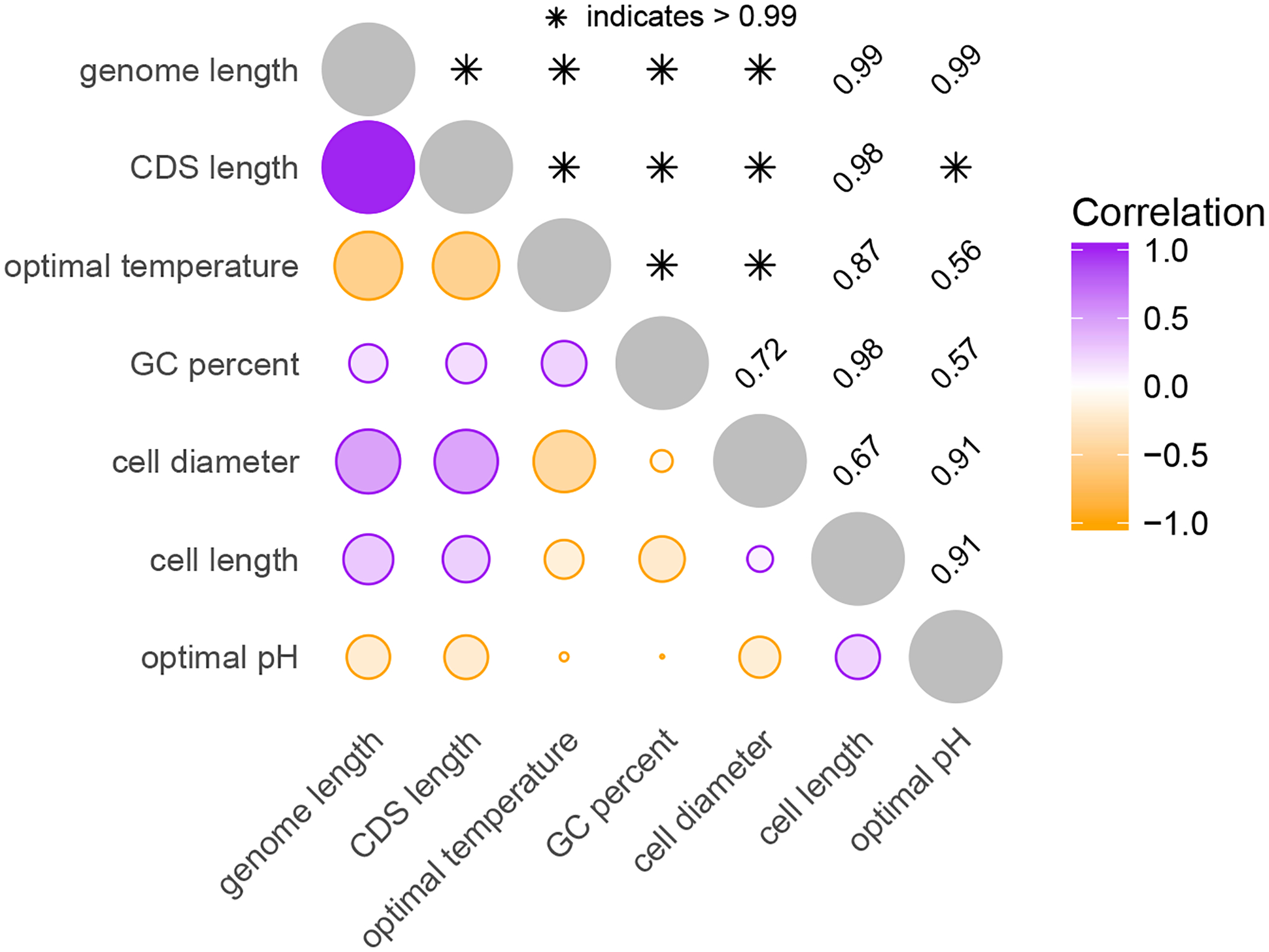
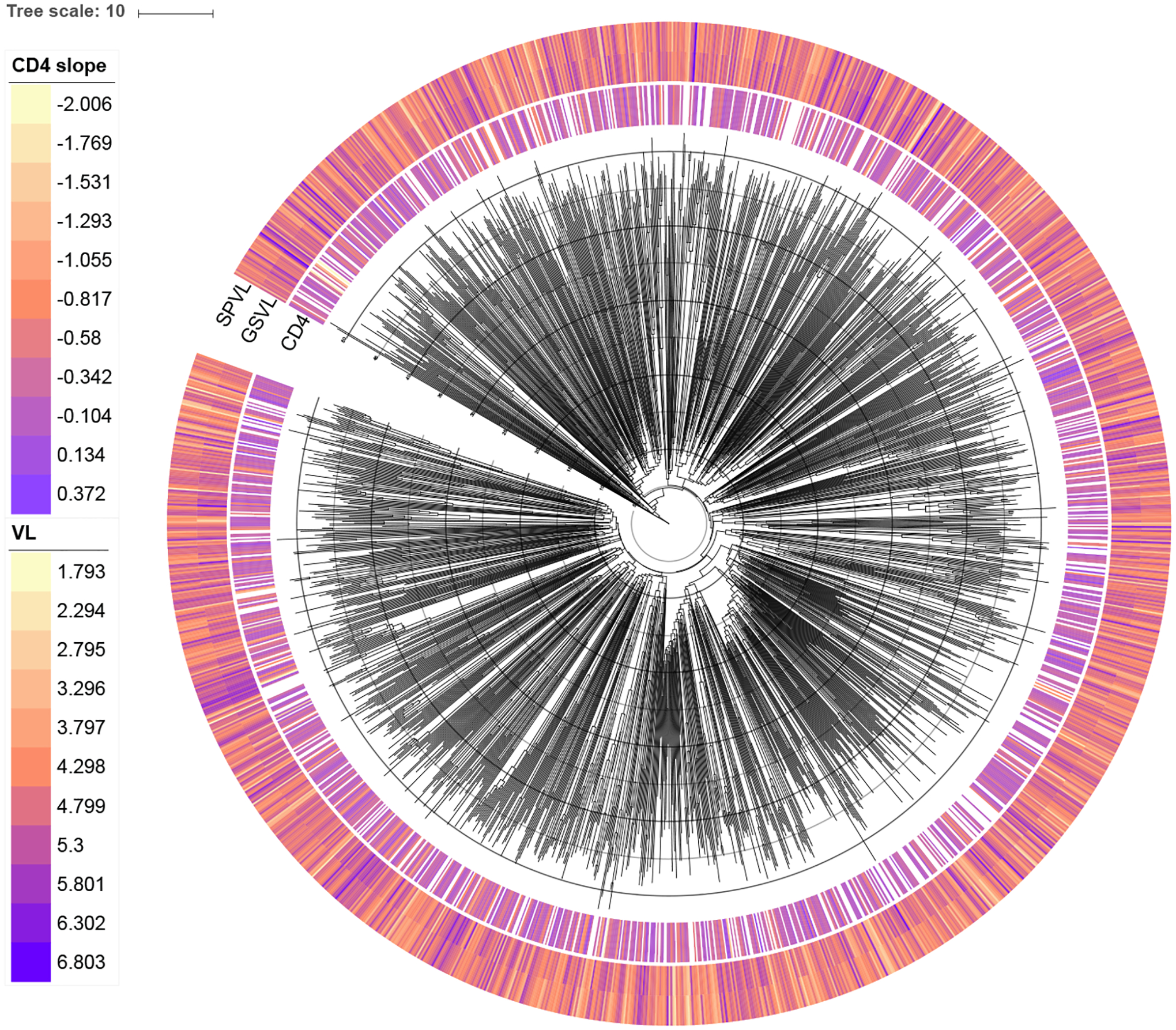
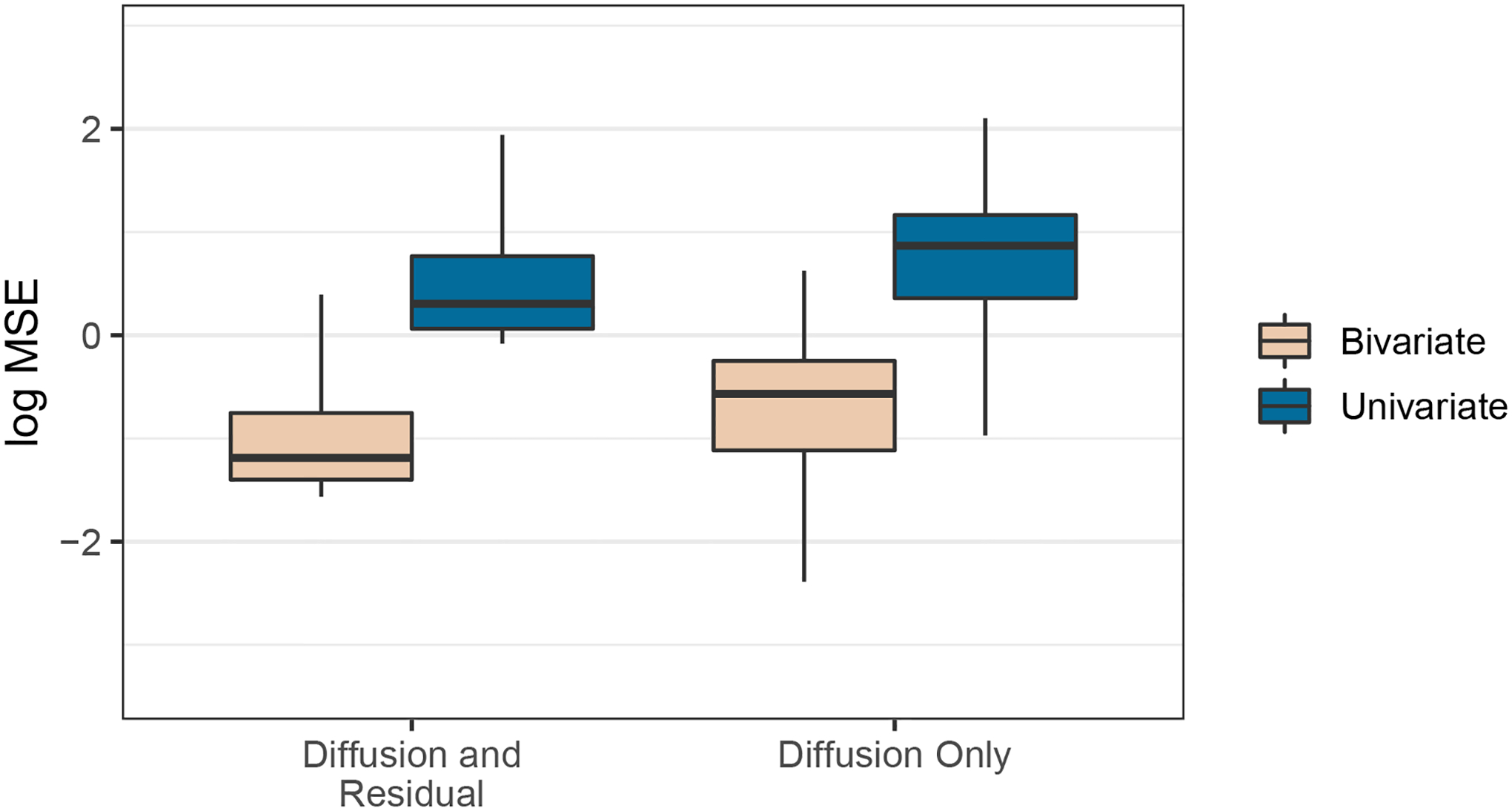
Comparative biologists are often interested in inferring covariation between multiple biological traits sampled across numerous related taxa. To properly study these relationships, we must control for the shared evolutionary history of the taxa to avoid spurious inference. An additional challenge arises as obtaining a full suite of measurements becomes increasingly difficult with increasing taxa. This generally necessitates data imputation or integration, and existing control techniques typically scale poorly as the number of taxa increases. We propose an inference technique that integrates out missing measurements analytically and scales linearly with the number of taxa by using a post-order traversal algorithm under a multivariate Brownian diffusion (MBD) model to characterize trait evolution. We further exploit this technique to extend the MBD model to account for sampling error or non-heritable residual variance. We test these methods to examine mammalian life history traits, prokaryotic genomic and phenotypic traits, and HIV infection traits. We find computational efficiency increases that top two orders-of-magnitude over current best practices. While we focus on the utility of this algorithm in phylogenetic comparative methods, our approach generalizes to solve long-standing challenges in computing the likelihood for matrix-normal and multivariate normal distributions with missing data at scale.
Keywords: Bayesian inference; matrix-normal; missing data; phylogenetics.
Figures

References
-
- Adams DC (2014). A method for assessing phylogenetic least squares models for shape and other high-dimensional multivariate data. Evolution 68(9), 2675–2688. - PubMed
-
- Alizon S, von Wyl V, Stadler T, Kouyos RD, Yerly S, Hirschel B, Böni J, Shah C, Klimkait T, Furrer H, Rauch A, Vernazza PL, Bernasconi E, Battegay M, Bürgisser P, Telenti A, Günthard HF, Bonhoeffer S, and Swiss HIV Cohort Study (2010, Sep). Phylogenetic approach reveals that virus genotype largely determines HIV set-point viral load. PLoS Pathogens 6(9), e1001123. - PMC - PubMed
-
- Bastide P, Ané C, Robin S, and Mariadassou M (2018). Inference of adaptive shifts for multivariate correlated traits. Systematic Biology 67(4), 662–680. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources