

Harnessing Water to Enhance Quadrupolar NMR Spectroscopy and Imaging
- PMID: 36062375
- PMCID: PMC9828088
- DOI: 10.1002/chem.202201490
Harnessing Water to Enhance Quadrupolar NMR Spectroscopy and Imaging
Abstract
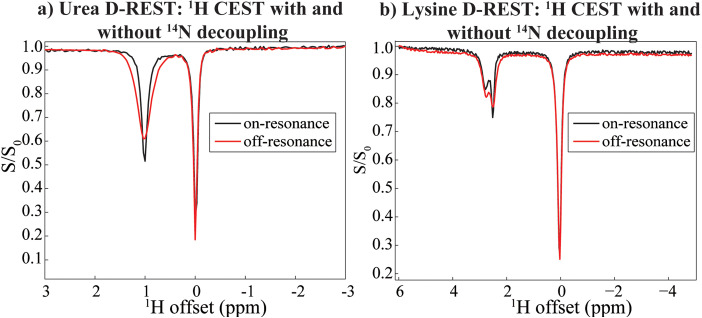
17 O and 14 N are attractive targets for in vivo NMR spectroscopy and imaging, but low gyromagnetic ratios γ and fast spin relaxation complicate observations. This work explores indirect ways of detecting some of these sites with the help of proton-detected double resonance techniques. As standard coherence transfer methods are of limited use for such indirect detection, alternative routes for probing the quadrupolar spectra on 1 H were tested. These centered on modulating the broadening effects imparted onto protons adjacent to the low-γ species through J couplings through either continuous wave or spin-echo double-resonance decoupling/recoupling sequences. As in all cases, the changes imparted by these double-resonance strategies were small due to the fast relaxation undergone by the quadrupoles, the sensitivity of these approaches was amplified by transferring their effects onto the abundant water 1 H signal. These amplifications were mediated by the spontaneous exchanges that the labile 1 Hs bound to 17 O or 14 N undergo with the water protons. In experiments designed on the basis of double-resonance spin echoes, these enhancements were imparted by looping the transverse encodings together with multiple longitudinal storage periods, leading to decoupling-recoupling with exchange (D-REX) sequences. In experiments designed on the basis of continuous on/off quadrupolar decoupling, these solvent exchanges were incorporated into chemical-exchange saturation transfer schemes, leading to decoupling-recoupling with saturation transfer (D-REST) sequences. Both of these variants harnessed sizable proportions of the easily detectable water signals, in order to characterize the NMR spectra and/or to image with atomic-site specificity the 17 O and 14 N species.
Keywords: chemical exchange; low-gamma MRI; quadrupolar NMR spectroscopy; sensitivity enhancement; water detection.
© 2022 The Authors. Chemistry - A European Journal published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures








Similar articles
-
Proton detection of MAS solid-state NMR spectra of half-integer quadrupolar nuclei.Solid State Nucl Magn Reson. 2017 Jul-Aug;84:171-181. doi: 10.1016/j.ssnmr.2017.03.005. Epub 2017 Mar 30. Solid State Nucl Magn Reson. 2017. PMID: 28392024
-
Magnetization transfer from protons to quadrupolar nuclei in solid-state NMR using PRESTO or dipolar-mediated refocused INEPT methods.J Magn Reson. 2019 Feb;299:109-123. doi: 10.1016/j.jmr.2018.12.016. Epub 2018 Dec 18. J Magn Reson. 2019. PMID: 30594000
-
Improving the resolution in proton-detected through-space heteronuclear multiple quantum correlation NMR spectroscopy.J Magn Reson. 2014 Aug;245:38-49. doi: 10.1016/j.jmr.2014.05.006. Epub 2014 May 21. J Magn Reson. 2014. PMID: 24929867
-
Proton-Based Ultrafast Magic Angle Spinning Solid-State NMR Spectroscopy.Acc Chem Res. 2017 Apr 18;50(4):1105-1113. doi: 10.1021/acs.accounts.7b00082. Epub 2017 Mar 29. Acc Chem Res. 2017. PMID: 28353338 Free PMC article. Review.
-
Magnetization Transfer Contrast and Chemical Exchange Saturation Transfer MRI. Features and analysis of the field-dependent saturation spectrum.Neuroimage. 2018 Mar;168:222-241. doi: 10.1016/j.neuroimage.2017.04.045. Epub 2017 Apr 21. Neuroimage. 2018. PMID: 28435103 Free PMC article. Review.
Cited by
-
The Environmental Impact of Medical Imaging Agents and the Roadmap to Sustainable Medical Imaging.Adv Sci (Weinh). 2025 Mar;12(9):e2404411. doi: 10.1002/advs.202404411. Epub 2025 Feb 4. Adv Sci (Weinh). 2025. PMID: 39905748 Free PMC article. Review.
References
-
- R. A. de Graaf, In Vivo NMR Spectroscopy: Principles and Techniques, 3rd ed., Wiley, 2019.
-
- J. Cavanagh, W. J. Fairbrother, A. G. Palmer III, M. Rance, N. J. Skelton, Protein NMR Spectroscopy, Elsevier, 2007.
-
- Pegg D. T., Doddrell D. M., Brooks W. M., Robin Bendall M., J. Magn. Reson. 1981, 44, 32–40.
-
- Eykyn T. R., Philp D. J., Kuchel P. W., Chem. Phys. Lett. 2003, 376, 732–736.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
