

Aβ plaques do not protect against HSV-1 infection in a mouse model of familial Alzheimer's disease, and HSV-1 does not induce Aβ pathology in a model of late onset Alzheimer's disease
- PMID: 36064300
- PMCID: PMC9836376
- DOI: 10.1111/bpa.13116
Aβ plaques do not protect against HSV-1 infection in a mouse model of familial Alzheimer's disease, and HSV-1 does not induce Aβ pathology in a model of late onset Alzheimer's disease
Abstract
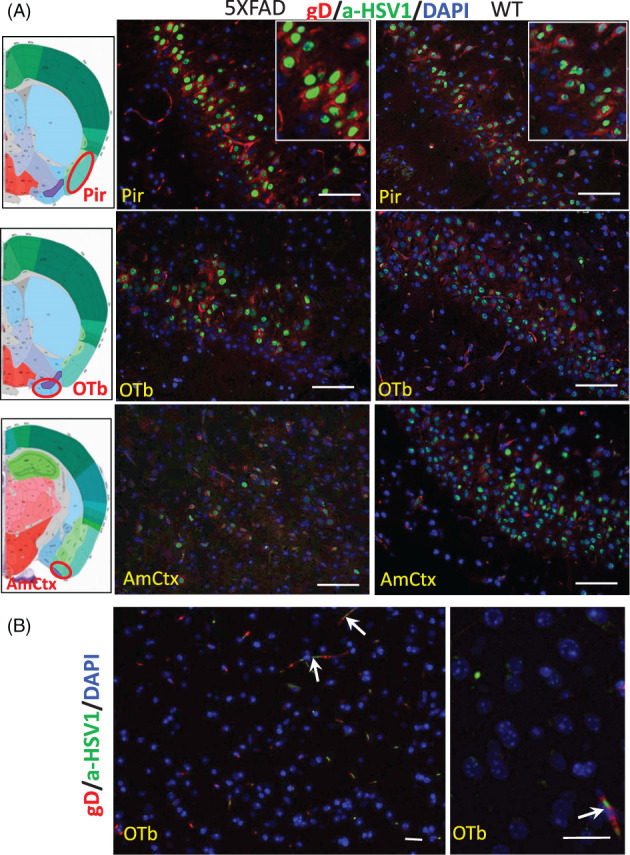
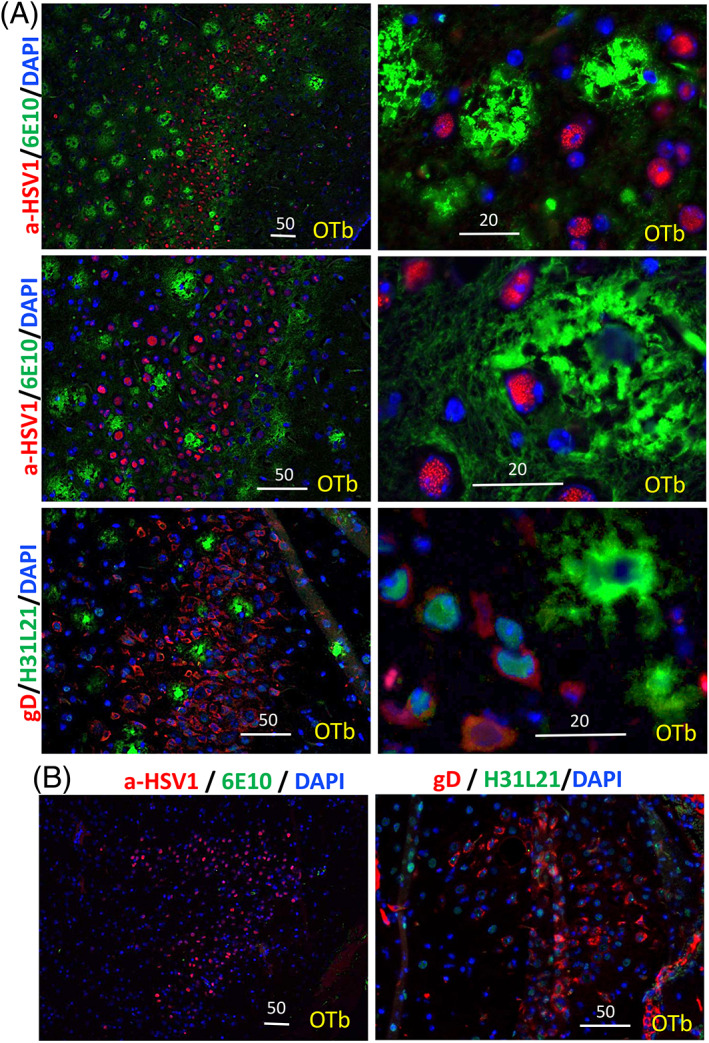
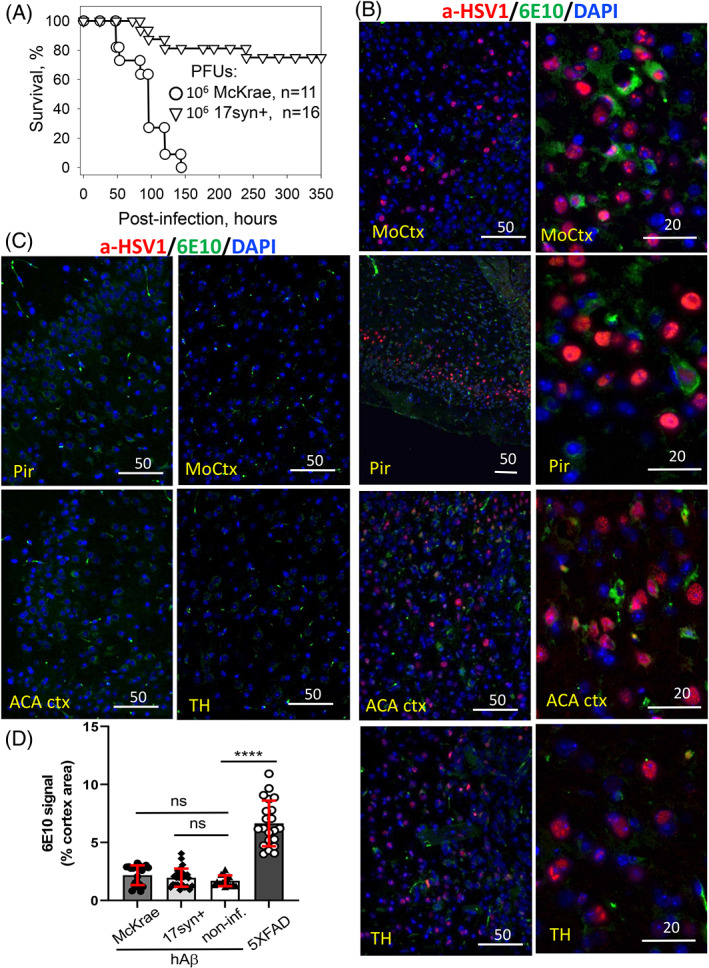
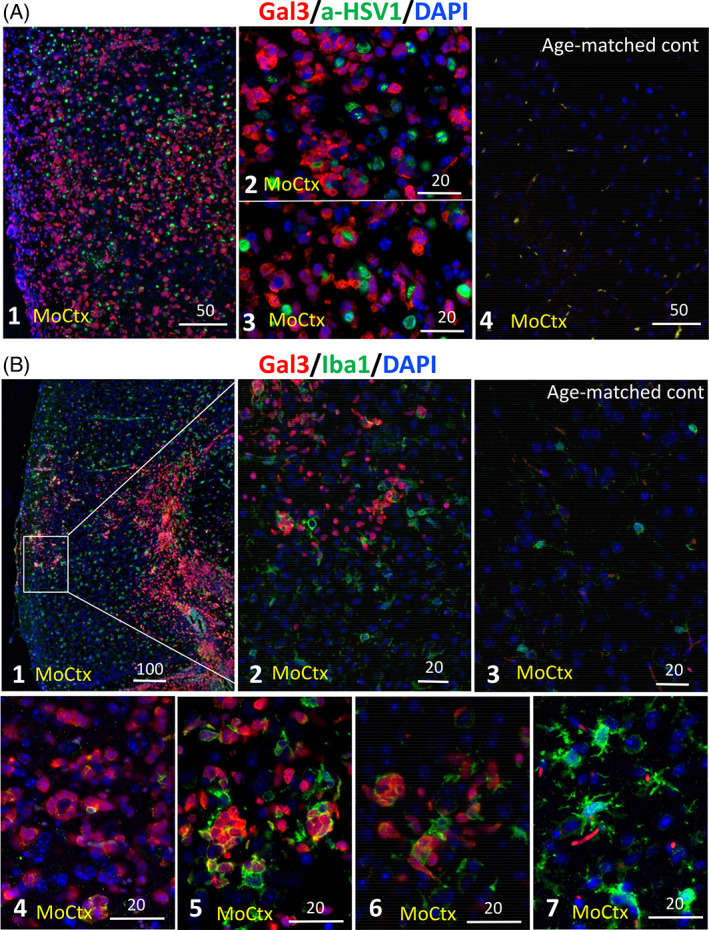
The possibility that the etiology of late onset Alzheimer's disease is linked to viral infections of the CNS has been actively debated in recent years. According to the antiviral protection hypothesis, viral pathogens trigger aggregation of Aβ peptides that are produced as a defense mechanism in response to infection to entrap and neutralize pathogens. To test the causative relationship between viral infection and Aβ aggregation, the current study examined whether Aβ plaques protect the mouse brain against Herpes Simplex Virus 1 (HSV-1) infection introduced via a physiological route and whether HSV-1 infection triggers formation of Aβ plaques in a mouse model of late-onset AD that does not develop Aβ pathology spontaneously. In aged 5XFAD mice infected via eye scarification, high density of Aβ aggregates did not improve survival time or rate when compared with wild type controls. In 5XFADs, viral replication sites were found in brain areas with a high density of extracellular Aβ deposits, however, no association between HSV-1 and Aβ aggregates could be found. To test whether HSV-1 triggers Aβ aggregation in a mouse model that lacks spontaneous Aβ pathology, 13-month-old hAβ/APOE4/Trem2*R47H mice were infected with HSV-1 via eye scarification with the McKrae HSV-1 strain, intracranial inoculation with McKrae, intracranial inoculation after priming with LPS for 6 weeks, or intracranial inoculation with high doses of McKrae or 17syn + strains that represent different degrees of neurovirulence. No signs of Aβ aggregation were found in any of the experimental groups. Instead, extensive infiltration of peripheral leukocytes was observed during the acute stage of HSV-1 infection, and phagocytic activity of myeloid cells was identified as the primary defense mechanism against HSV-1. The current results argue against a direct causative relationship between HSV-1 infection and Aβ pathology.
Keywords: 5XFAD mice; Alzheimer's disease; Aβ aggregates; amyloid precursor protein; hAβ/APOE4/Trem2*R47H mice; herpes simplex virus 1; infiltrating myeloid cells; microglia.
© 2022 The Authors. Brain Pathology published by John Wiley & Sons Ltd on behalf of International Society of Neuropathology.
Conflict of interest statement
The authors declare no conflict of interests.
Figures








References
-
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–90. - PubMed
-
- Goate A, Chartier‐Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–6. - PubMed
-
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–5. - PubMed
-
- Itzhaki RF, Wozniak MA, Appelt DM, Balin BJ. Infiltration of the brain by pathogens causes Alzheimer's disease. Neurobiol Aging. 2004;25(5):619–27. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
