

BREEDIT: a multiplex genome editing strategy to improve complex quantitative traits in maize
- PMID: 36066192
- PMCID: PMC9806654
- DOI: 10.1093/plcell/koac243
BREEDIT: a multiplex genome editing strategy to improve complex quantitative traits in maize
Erratum in
-
Correction to: BREEDIT: a multiplex genome editing strategy to improve complex quantitative traits in maize.Plant Cell. 2023 Mar 15;35(3):1160. doi: 10.1093/plcell/koad001. Plant Cell. 2023. PMID: 36683260 Free PMC article. No abstract available.
Abstract
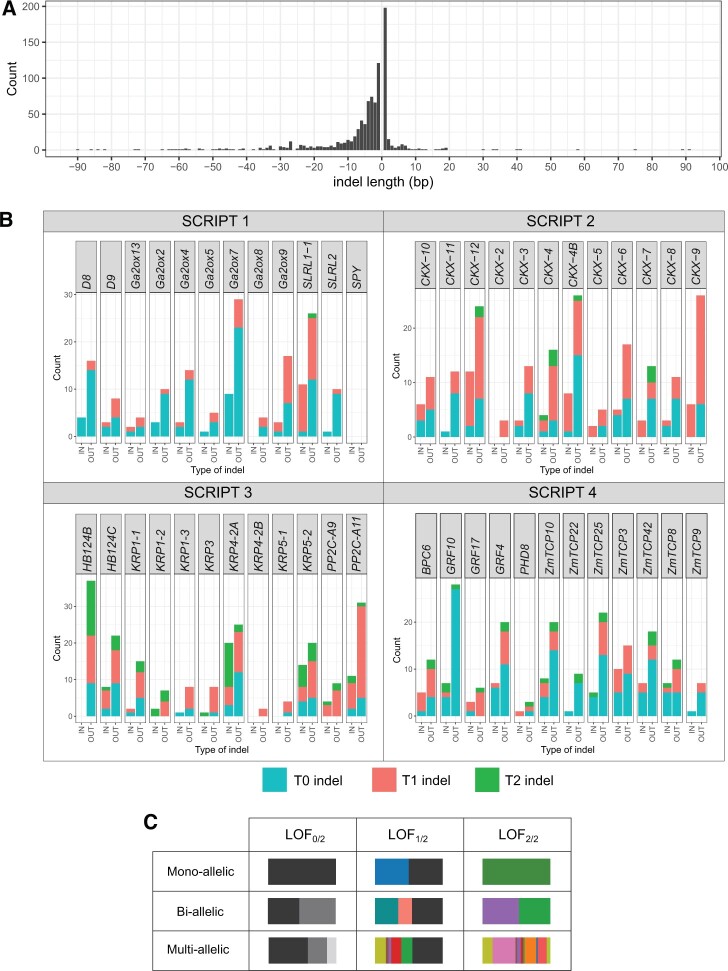
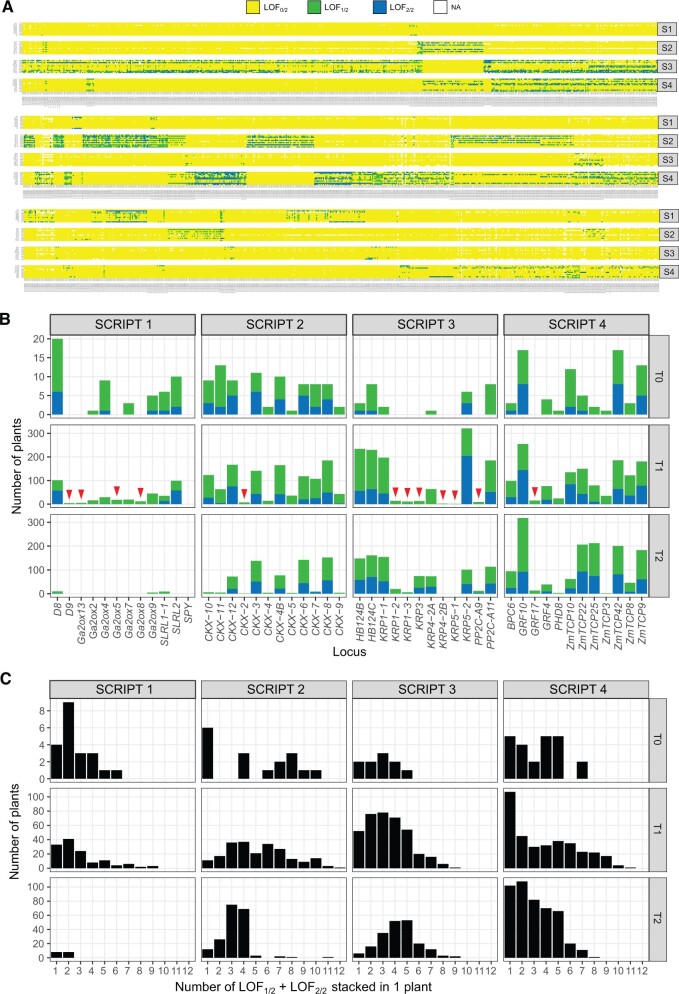
Ensuring food security for an ever-growing global population while adapting to climate change is the main challenge for agriculture in the 21st century. Although new technologies are being applied to tackle this problem, we are approaching a plateau in crop improvement using conventional breeding. Recent advances in CRISPR/Cas9-mediated gene engineering have paved the way to accelerate plant breeding to meet this increasing demand. However, many traits are governed by multiple small-effect genes operating in complex interactive networks. Here, we present the gene discovery pipeline BREEDIT, which combines multiplex genome editing of whole gene families with crossing schemes to improve complex traits such as yield and drought tolerance. We induced gene knockouts in 48 growth-related genes into maize (Zea mays) using CRISPR/Cas9 and generated a collection of over 1,000 gene-edited plants. The edited populations displayed (on average) 5%-10% increases in leaf length and up to 20% increases in leaf width compared with the controls. For each gene family, edits in subsets of genes could be associated with enhanced traits, allowing us to reduce the gene space to be considered for trait improvement. BREEDIT could be rapidly applied to generate a diverse collection of mutants to identify promising gene modifications for later use in breeding programs.
© American Society of Plant Biologists 2022. All rights reserved. For permissions, please email: journals.permissions@oup.com.
Figures





Comment in
-
BREEDIT: Fast breeding tools to match the fast pace of climate change.Plant Cell. 2023 Jan 2;35(1):4-5. doi: 10.1093/plcell/koac328. Plant Cell. 2023. PMID: 36433789 Free PMC article. No abstract available.
References
-
- Aesaert S, Impens L, Coussens G, Van Lerberge E, Vanderhaeghen R, Desmet L, Vanhevel Y, Bossuyt S, Wambua AN, Van Lijsebettens M, et al. (2022) Optimized transformation and gene editing of the B104 public maize inbred by improved tissue culture and use of morphogenic regulators. Front Plant Sci 13: 883847. - PMC - PubMed
-
- Anzalone AV, Koblan LW, Liu DR (2020) Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol 38: 824–844 - PubMed
-
- Ashikari M, Sakakibara H, Lin S, Yamamoto T, Takashi T, Nishimura A, Angeles ER, Qian Q, Kitano H, Matsuoka M (2005) Cytokinin oxidase regulates rice grain production. Science 309: 741–745 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
