

Germline homozygous missense DEPDC5 variants cause severe refractory early-onset epilepsy, macrocephaly and bilateral polymicrogyria
- PMID: 36067010
- PMCID: PMC9896472
- DOI: 10.1093/hmg/ddac225
Germline homozygous missense DEPDC5 variants cause severe refractory early-onset epilepsy, macrocephaly and bilateral polymicrogyria
Abstract
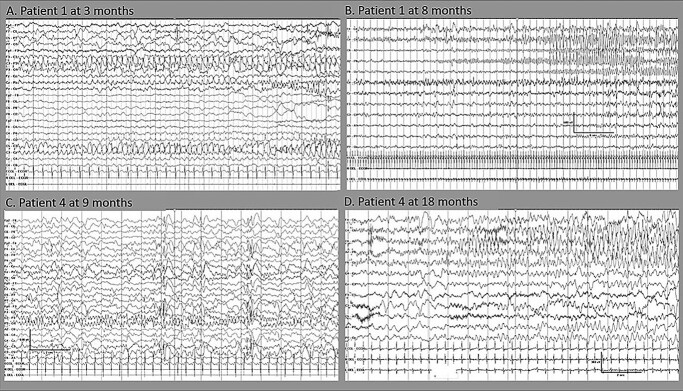
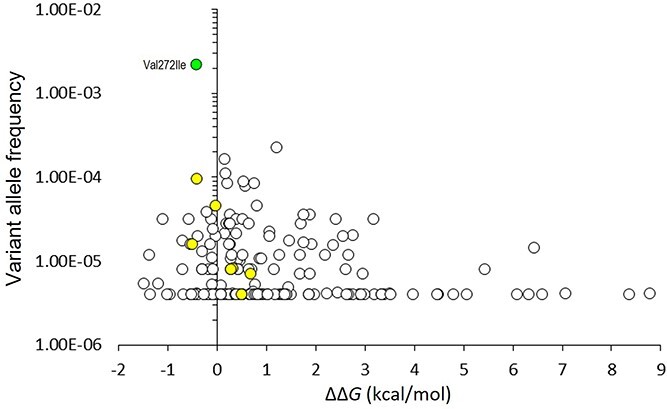
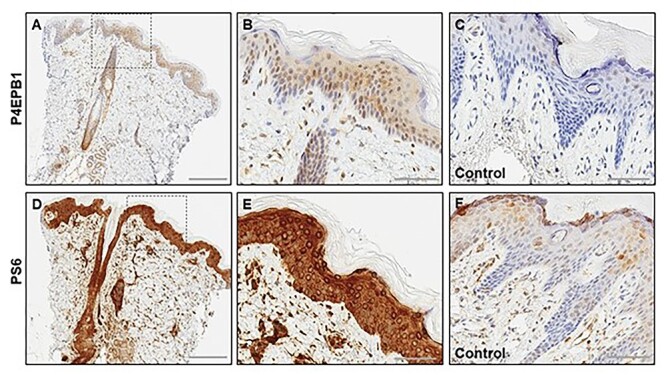
DEPDC5 (DEP Domain-Containing Protein 5) encodes an inhibitory component of the mammalian target of rapamycin (mTOR) pathway and is commonly implicated in sporadic and familial focal epilepsies, both non-lesional and in association with focal cortical dysplasia. Germline pathogenic variants are typically heterozygous and inactivating. We describe a novel phenotype caused by germline biallelic missense variants in DEPDC5. Cases were identified clinically. Available records, including magnetic resonance imaging and electroencephalography, were reviewed. Genetic testing was performed by whole exome and whole-genome sequencing and cascade screening. In addition, immunohistochemistry was performed on skin biopsy. The phenotype was identified in nine children, eight of which are described in detail herein. Six of the children were of Irish Traveller, two of Tunisian and one of Lebanese origin. The Irish Traveller children shared the same DEPDC5 germline homozygous missense variant (p.Thr337Arg), whereas the Lebanese and Tunisian children shared a different germline homozygous variant (p.Arg806Cys). Consistent phenotypic features included extensive bilateral polymicrogyria, congenital macrocephaly and early-onset refractory epilepsy, in keeping with other mTOR-opathies. Eye and cardiac involvement and severe neutropenia were also observed in one or more patients. Five of the children died in infancy or childhood; the other four are currently aged between 5 months and 6 years. Skin biopsy immunohistochemistry was supportive of hyperactivation of the mTOR pathway. The clinical, histopathological and genetic evidence supports a causal role for the homozygous DEPDC5 variants, expanding our understanding of the biology of this gene.
© The Author(s) 2022. Published by Oxford University Press.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
