

IRF3 inhibits nuclear translocation of NF-κB to prevent viral inflammation
- PMID: 36067309
- PMCID: PMC9478676
- DOI: 10.1073/pnas.2121385119
IRF3 inhibits nuclear translocation of NF-κB to prevent viral inflammation
Abstract
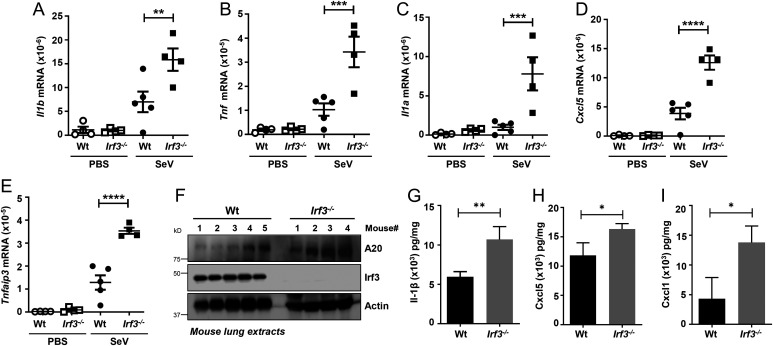
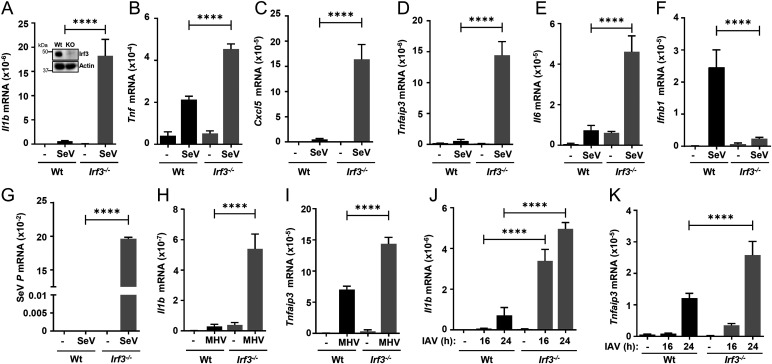
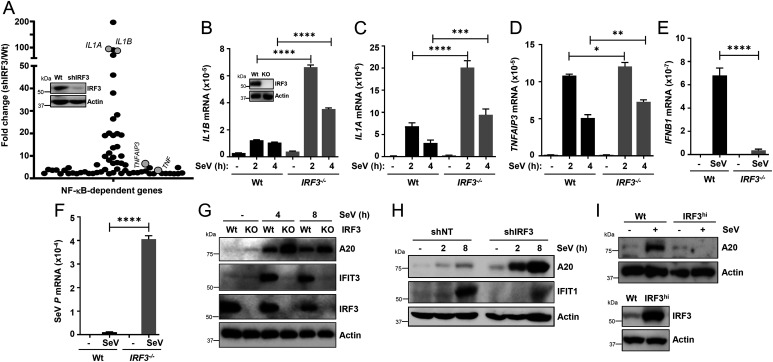
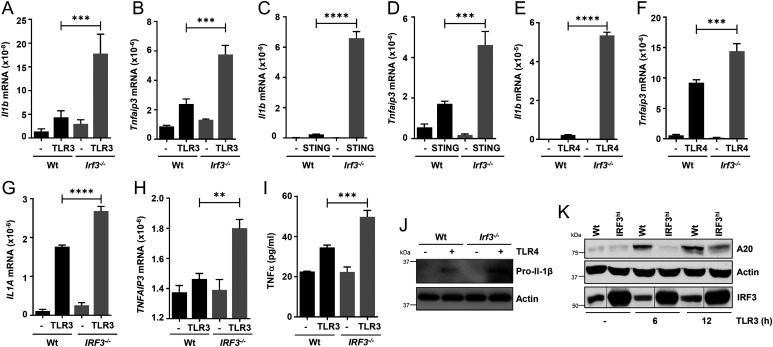
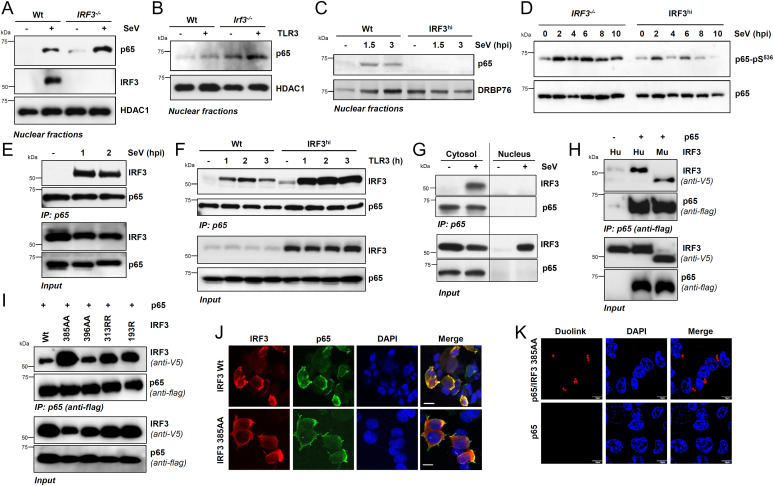
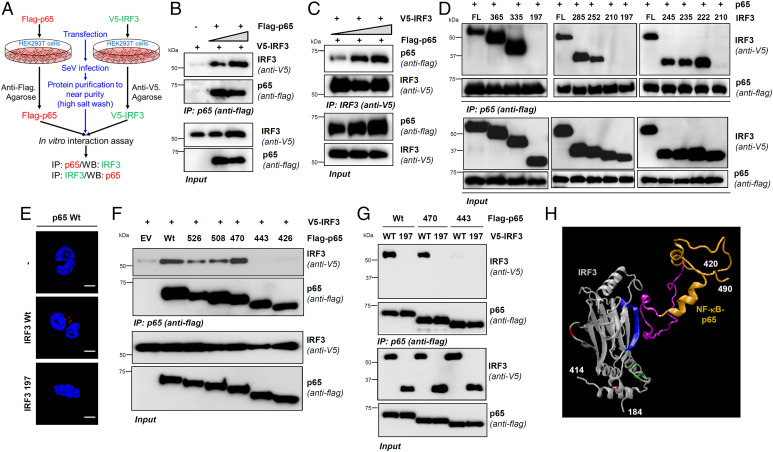
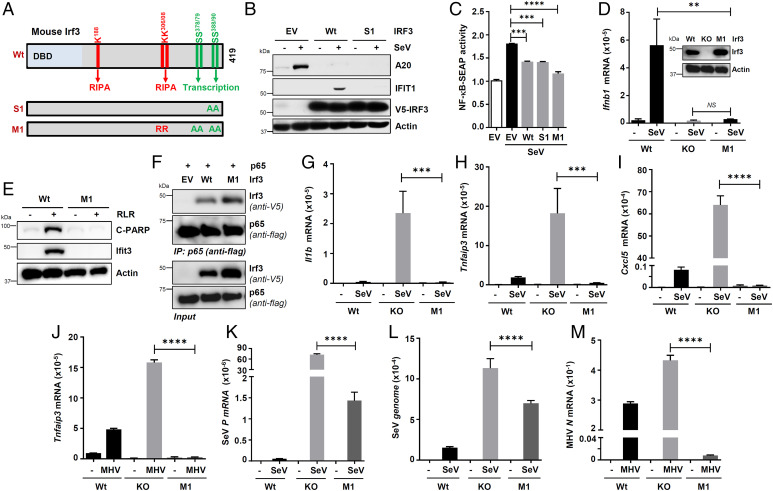
Interferon (IFN) regulatory factor 3 (IRF3) is a transcription factor activated by phosphorylation in the cytoplasm of a virus-infected cell; by translocating to the nucleus, it induces transcription of IFN-β and other antiviral genes. We have previously reported IRF3 can also be activated, as a proapoptotic factor, by its linear polyubiquitination mediated by the RIG-I pathway. Both transcriptional and apoptotic functions of IRF3 contribute to its antiviral effect. Here, we report a nontranscriptional function of IRF3, namely, the repression of IRF3-mediated NF-κB activity (RIKA), which attenuated viral activation of NF-κB and the resultant inflammatory gene induction. In Irf3-/- mice, consequently, Sendai virus infection caused enhanced inflammation in the lungs. Mechanistically, RIKA was mediated by the direct binding of IRF3 to the p65 subunit of NF-κB in the cytoplasm, which prevented its nuclear import. A mutant IRF3 defective in both the transcriptional and the apoptotic activities was active in RIKA and inhibited virus replication. Our results demonstrated IRF3 deployed a three-pronged attack on virus replication and the accompanying inflammation.
Keywords: IRF3; NF-κB; antiviral; innate immunity; viral inflammation.
Conflict of interest statement
The authors declare no competing interest.
Figures
Similar articles
-
PKC alpha regulates Sendai virus-mediated interferon induction through HDAC6 and β-catenin.EMBO J. 2011 Sep 27;30(23):4838-49. doi: 10.1038/emboj.2011.351. EMBO J. 2011. PMID: 21952047 Free PMC article.
-
Japanese Encephalitis Virus NS5 Inhibits Type I Interferon (IFN) Production by Blocking the Nuclear Translocation of IFN Regulatory Factor 3 and NF-κB.J Virol. 2017 Mar 29;91(8):e00039-17. doi: 10.1128/JVI.00039-17. Print 2017 Apr 15. J Virol. 2017. PMID: 28179530 Free PMC article.
-
Induction of INKIT by Viral Infection Negatively Regulates Antiviral Responses through Inhibiting Phosphorylation of p65 and IRF3.Cell Host Microbe. 2017 Jul 12;22(1):86-98.e4. doi: 10.1016/j.chom.2017.06.013. Cell Host Microbe. 2017. PMID: 28704656
-
RIG-I-like receptor-induced IRF3 mediated pathway of apoptosis (RIPA): a new antiviral pathway.Protein Cell. 2017 Mar;8(3):165-168. doi: 10.1007/s13238-016-0334-x. Epub 2016 Nov 4. Protein Cell. 2017. PMID: 27815826 Free PMC article. Review.
-
Transcriptional and Non-Transcriptional Activation, Posttranslational Modifications, and Antiviral Functions of Interferon Regulatory Factor 3 and Viral Antagonism by the SARS-Coronavirus.Viruses. 2021 Mar 29;13(4):575. doi: 10.3390/v13040575. Viruses. 2021. PMID: 33805458 Free PMC article. Review.
Cited by
-
Pseudorabies virus VHS protein abrogates interferon responses by blocking NF-κB and IRF3 nuclear translocation.Virol Sin. 2024 Aug;39(4):587-599. doi: 10.1016/j.virs.2024.05.009. Epub 2024 May 30. Virol Sin. 2024. PMID: 38823782 Free PMC article.
-
How Different Pathologies Are Affected by IFIT Expression.Viruses. 2023 Jan 25;15(2):342. doi: 10.3390/v15020342. Viruses. 2023. PMID: 36851555 Free PMC article. Review.
-
Helicobacter pylori infection induces DNA double-strand breaks through the ACVR1/IRF3/POLD1 signaling axis to drive gastric tumorigenesis.Gut Microbes. 2025 Dec;17(1):2463581. doi: 10.1080/19490976.2025.2463581. Epub 2025 Feb 9. Gut Microbes. 2025. PMID: 39924917 Free PMC article.
-
Susceptibility to Zika virus in a Collaborative Cross mouse strain is induced by Irf3 deficiency in vitro but requires other variants in vivo.PLoS Pathog. 2023 Sep 21;19(9):e1011446. doi: 10.1371/journal.ppat.1011446. eCollection 2023 Sep. PLoS Pathog. 2023. PMID: 37733807 Free PMC article.
-
Innate immune role of IL-6 in influenza a virus pathogenesis.Front Cell Infect Microbiol. 2025 Jul 7;15:1605446. doi: 10.3389/fcimb.2025.1605446. eCollection 2025. Front Cell Infect Microbiol. 2025. PMID: 40692679 Free PMC article. Review.
References
-
- White C. L., Sen G. C., “Interferons and antiviral actions” inCellular Signaling and Innate Immune Responses to RNA Virus Infections, Brasier A. R., Garcia-Sastre A., Lemon S. M., Eds. (ASM Press, Washington, DC, 2008), pp. 91–106.
-
- Biron C. A., Sen G. C., “Innate responses to viral infections” in Fields Virology, Knipe D. M., Howley P. M., Eds. (Lippincott, Williams and Wilkins, Philadelphia, PA, ed. 5, 2006), pp. 249–278.
-
- Fensterl V., Sen G. C., Interferons and viral infections. Biofactors 35, 14–20 (2009). - PubMed
-
- Schoggins J. W., Interferon-stimulated genes: What do they all do? Annu. Rev. Virol. 6, 567–584 (2019). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
